Summary: According to a new study, a drug used to treat multiple sclerosis could help to treat pain caused by cancer treatments in myeloma patients.
Source: Rockefeller University Press.
Researchers from the Saint Louis University School of Medicine have discovered why many multiple myeloma patients experience severe pain when treated with the anticancer drug bortezomib. The study, which will be published April 27 in the Journal of Experimental Medicine, suggests that a drug already approved to treat multiple sclerosis could mitigate this effect, allowing myeloma patients to successfully complete their treatment and relieving the pain of myeloma survivors.
Chemotherapy-induced peripheral neuropathy (CIPN) is a common, painful side effect of many anticancer drugs that can cause patients to discontinue treatment or, because symptoms can persist for years, reduce the quality of life for cancer survivors. “This growing problem is a major unmet clinical need because the increased efficacy of cancer therapy has resulted in nearly 14 million cancer survivors in the United States, many suffering from the long-term side effects of CIPN,” says Daniela Salvemini, Professor of Pharmacology and Physiology at the Saint Louis University School of Medicine.
Bortezomib, which is widely used to treat multiple myeloma and mantle cell lymphoma, causes CIPN in over 40% of patients, but the reasons for this are unclear. Salvemini and colleagues found that bortezomib accelerates the production of a class of molecules called sphingolipids that have previously been linked to neuropathic pain. Rats treated with bortezomib began to accumulate two sphingolipid metabolites, sphingosine 1-phosphate and dihydrosphingosine 1-phosphate, in their spinal cords at the time that they began to show signs of neuropathic pain. Blocking the production of these molecules prevented the animals from developing CIPN in response to bortezomib.
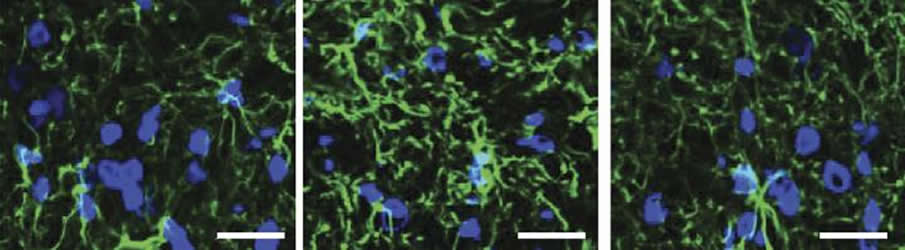
Sphingosine 1-phosphate and dihydrosphingosine 1-phosphate can activate a cell surface receptor protein called S1PR1. Salvemini and colleagues determined that the two metabolites cause CIPN by activating S1PR1 on the surface of specialized nervous system support cells called astrocytes, resulting in neuroinflammation and enhanced release of the excitatory neurotransmitter glutamate.
Drugs that inhibit S1PR1 also prevented rats from developing CIPN in response to bortezomib. One such inhibitor was fingolimod, an orally administered drug approved to treat multiple sclerosis. Importantly, fingolimod did not inhibit bortezomib’s ability to kill myeloma cells. Indeed, fingolimod itself has been reported to inhibit tumor growth and enhance the effects of bortezomib. “Because fingolimod shows promising anticancer potential and is already FDA approved, we think that our findings in rats can be rapidly translated to the clinic to prevent and treat bortezomib-induced neuropathic pain,” Salvemini says.
Funding: This work was funded by the Mayday Fund, Leukemia and Lymphoma Society, National Institutes of Health, NIH/National Cancer Institute,.
Source: Ben Short – Rockefeller University Press
Publisher: Organized by NeuroscienceNews.com.
Image Source: NeuroscienceNews.com image is credited to Stockstill et al., 2018.
Original Research: Open access research for “Dysregulation of sphingolipid metabolism contributes to bortezomib-induced neuropathic pain” by Katherine Stockstill,Timothy M. Doyle, Xisheng Yan, Zhoumou Chen, Kali Janes, Joshua W. Little, Kathryn Braden, Filomena Lauro, Luigino Antonio Giancotti,Caron Mitsue Harada, Ruchi Yadav, Wen Hua Xiao,Jack M. Lionberger,William L. Neumann, Gary J. Bennett, Han-Rong Weng, Sarah Spiegel, and Daniela Salvemini in Journal of Experimental Medicine. Published April 27 2018.
doi:10.1084/jem.20170584
[cbtabs][cbtab title=”MLA”]Rockefeller University Press “Multiple Sclerosis Drug Could Reduce Painful Side Effects of Common Cancer Treatment.” NeuroscienceNews. NeuroscienceNews, 25 April 2018.
<https://neurosciencenews.com/ms-drug-pain-cancer-8898/>.[/cbtab][cbtab title=”APA”]Rockefeller University Press (2018, April 25). Multiple Sclerosis Drug Could Reduce Painful Side Effects of Common Cancer Treatment. NeuroscienceNews. Retrieved April 25, 2018 from https://neurosciencenews.com/ms-drug-pain-cancer-8898/[/cbtab][cbtab title=”Chicago”]Rockefeller University Press “Multiple Sclerosis Drug Could Reduce Painful Side Effects of Common Cancer Treatment.” https://neurosciencenews.com/ms-drug-pain-cancer-8898/ (accessed April 25, 2018).[/cbtab][/cbtabs]
Abstract
Dysregulation of sphingolipid metabolism contributes to bortezomib-induced neuropathic pain
The development of chemotherapy-induced painful peripheral neuropathy is a major dose-limiting side effect of many chemotherapeutics, including bortezomib, but the mechanisms remain poorly understood. We now report that bortezomib causes the dysregulation of de novo sphingolipid metabolism in the spinal cord dorsal horn to increase the levels of sphingosine-1-phosphate (S1P) receptor 1 (S1PR1) ligands, S1P and dihydro-S1P. Accordingly, genetic and pharmacological disruption of S1PR1 with multiple S1PR1 antagonists, including FTY720, blocked and reversed neuropathic pain. Mice with astrocyte-specific alterations of S1pr1 did not develop neuropathic pain and lost their ability to respond to S1PR1 inhibition, strongly implicating astrocytes as a primary cellular substrate for S1PR1 activity. At the molecular level, S1PR1 engaged astrocyte-driven neuroinflammation and altered glutamatergic homeostasis, processes blocked by S1PR1 antagonism. Our findings establish S1PR1 as a target for therapeutic intervention and provide insight into cellular and molecular pathways. As FTY720 also shows promising anticancer potential and is FDA approved, rapid clinical translation of our findings is anticipated.