Summary: Researchers have identified one way in which an RNA binding protein may contribute to ALS.
Source: Case Western Reserve.
Findings may have implications for Lou Gehrig’s, Alzheimer’s, Parkinson’s and Huntington’s diseases.
Amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease) is a progressive disorder that devastates motor nerve cells. People diagnosed with ALS slowly lose the ability to control muscle movement, and are ultimately unable to speak, eat, move, or breathe. The cellular mechanisms behind ALS are also found in certain types of dementia.
A groundbreaking scientific study published in Nature Medicine has found one way an RNA binding protein may contribute to ALS disease progression. Cells make RNA to carry instructions for making proteins from DNA to protein-constructing machinery.
The culprit protein, TDP-43, normally binds to small pieces of newly read RNA and helps shuttle the fragments around inside nerve cell nuclei. The study describes for the first time the molecular consequences of misplaced TDP-43 inside nerve cells, and demonstrates that correcting its location can restore nerve cell function. Misplacement of TDP-43 in nerve cells is a hallmark of ALS and other neurological disorders including frontotemporal dementia (FTD), Alzheimer’s, Parkinson’s, and Huntington’s diseases. Studies that characterize common mechanisms behind these diseases could have widespread implications and may also accelerate development of broad-based therapies.
To find the misplaced TDP-43, the researchers viewed nerve cells donated by people who died from ALS or FTD under high powered microscopes. They discovered TDP-43 accumulates in nerve cell mitochondria, critical structures responsible for generating the enormous amount of energy nerve cells require. By physically isolating the affected mitochondria the researchers were able to pinpoint TDP-43’s exact location inside the subcellular structures. They were also able to characterize variations of the protein most likely to get misplaced.
This important work was led by Xinglong Wang, PhD, from the department of pathology at Case Western Reserve University School of Medicine and a team of scientists from his laboratory.
“By multiple approaches, we have identified the mitochondrial inner membrane facing matrix as the major site for mitochondrial TDP-43,” explained Wang. “Mitochondria might be major accumulation sites of TDP-43 in dying neurons in various major neurodegenerative diseases.”
The researchers discovered that once inside the mitochondria, TDP-43 resumes its RNA binding role and attaches itself to mitochondrial genetic material. This disrupts the mitochondria’s ability to generate energy for the cell. Wang’s team was able to precisely identify the RNA in mitochondria that was bound by TDP-43 and observe the resultant disassembly of mitochondrial protein complexes. This finding provides much needed clarity on the consequences of TDP-43 misplacement inside nerve cells and opens the door for deeper studies involving a range of neurological disorders. Although the study focused on ALS and FTD, according to Wang “mislocalization of TDP-43 represents a key pathological feature correlating strongly with symptoms in more than half of Alzheimer’s disease patients.”
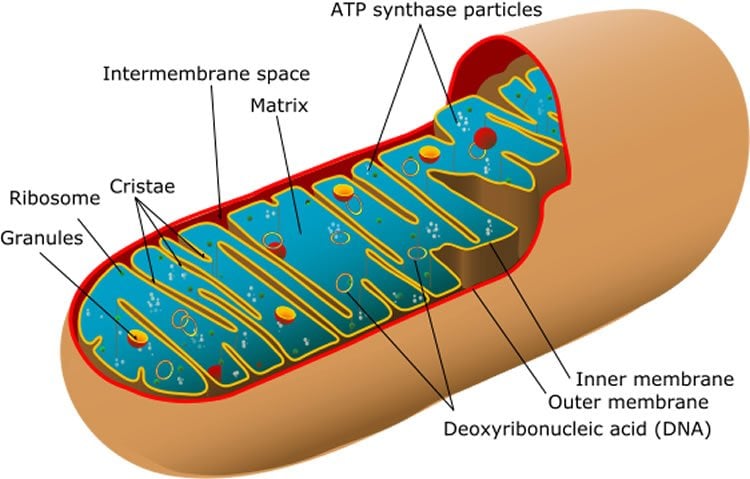
Mutations in the gene encoding TDP-43 have long been linked to neurodegenerative diseases like ALS and FTD. Wang’s team found that disease-associated mutations in TDP-43 enhance its misplacement inside nerve cells. The researchers also identified sections of TDP-43 that are recognized by mitochondria and serve as signals to let it inside. These sections could serve as therapeutic targets, as the study found blocking them prevents TDP-43 from localizing inside mitochondria. Importantly, Wang’s team was able to keep TDP-43 out of nerve cell mitochondria in mice using small proteins which “almost completely” prevented nerve cell toxicity and disease progression.
“We, for the first time, provide the novel concept that the inhibition of TDP-43 mitochondrial localization is sufficient to prevent TDP-43-linked neurodegeneration,” said Wang. “Targeting mitochondrial TDP-43 could be a novel therapeutic approach for ALS, FTD and other TDP-43-linked neurodegenerative diseases.”
Wang has begun to develop small proteins that prevent TDP-43 from reaching mitochondria in human nerve cells, and has a patent pending for the therapeutic molecule used in the study.
There is no treatment currently available for ALS or FTD. The average life expectancy for people newly diagnosed with ALS is just three years, according to The ALS Association.
Funding: This work was supported by grants from the National Institutes of Health (R03AG044680 and 1R01NS089604), the Alzheimer’s Association (2014-NIRG-301299) and the University Hospitals of Cleveland (2012 SPITZ Innovation Pilot Grant).
Source: Jay Shah – Case Western Reserve
Image Source: This NeuroscienceNews.com image is in the public domain.
Original Research: Abstract for “The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity” by Wenzhang Wang, Luwen Wang, Junjie Lu, Sandra L Siedlak, Hisashi Fujioka, Jingjing Liang, Sirui Jiang, Xiaopin Ma, Zhen Jiang, Edroaldo Lummertz da Rocha, Max Sheng, Heewon Choi, Paul H Lerou, Hu Li and Xinglong Wang in Nature Medicine. Published online June 27 2016 doi:10.1038/nm.4130
[cbtabs][cbtab title=”MLA”]Case Western Reserve. “Scientists Keep a Molecule from Moving Inside Nerve Cells to Prevent Cell Death.” NeuroscienceNews. NeuroscienceNews, 3 August 2016.
<https://neurosciencenews.com/molecule-neuron-apoptosis-4773/>.[/cbtab][cbtab title=”APA”]Case Western Reserve. (2016, August 3). Scientists Keep a Molecule from Moving Inside Nerve Cells to Prevent Cell Death. NeuroscienceNew. Retrieved August 3, 2016 from https://neurosciencenews.com/molecule-neuron-apoptosis-4773/[/cbtab][cbtab title=”Chicago”]Case Western Reserve. “Scientists Keep a Molecule from Moving Inside Nerve Cells to Prevent Cell Death.” https://neurosciencenews.com/molecule-neuron-apoptosis-4773/ (accessed August 3, 2016).[/cbtab][/cbtabs]
Abstract
The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity
Genetic mutations in TAR DNA-binding protein 43 (TARDBP, also known as TDP-43) cause amyotrophic lateral sclerosis (ALS), and an increase in the presence of TDP-43 (encoded by TARDBP) in the cytoplasm is a prominent histopathological feature of degenerating neurons in various neurodegenerative diseases. However, the molecular mechanisms by which TDP-43 contributes to ALS pathophysiology remain elusive. Here we have found that TDP-43 accumulates in the mitochondria of neurons in subjects with ALS or frontotemporal dementia (FTD). Disease-associated mutations increase TDP-43 mitochondrial localization. In mitochondria, wild-type (WT) and mutant TDP-43 preferentially bind mitochondria-transcribed messenger RNAs (mRNAs) encoding respiratory complex I subunits ND3 and ND6, impair their expression and specifically cause complex I disassembly. The suppression of TDP-43 mitochondrial localization abolishes WT and mutant TDP-43-induced mitochondrial dysfunction and neuronal loss, and improves phenotypes of transgenic mutant TDP-43 mice. Thus, our studies link TDP-43 toxicity directly to mitochondrial bioenergetics and propose the targeting of TDP-43 mitochondrial localization as a promising therapeutic approach for neurodegeneration.
“The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity” by Wenzhang Wang, Luwen Wang, Junjie Lu, Sandra L Siedlak, Hisashi Fujioka, Jingjing Liang, Sirui Jiang, Xiaopin Ma, Zhen Jiang, Edroaldo Lummertz da Rocha, Max Sheng, Heewon Choi, Paul H Lerou, Hu Li and Xinglong Wang in Nature Medicine. Published online June 27 2016 doi:10.1038/nm.4130