Summary: Researchers have generated motor neurons from people with familial ALS who carry FUS mutations.
Source: VIB Flanders.
A team of researchers at VIB and KU Leuven used stem cell technology to generate motor neurons from ALS patients carrying mutations in FUS. They found disturbed axonal transport in these motor neurons, but also identified genetic and pharmacological strategies that mitigate this defect.
Amyotrophic lateral sclerosis (ALS) is a deadly, incurable neurodegenerative disorder. Patients experience progressive paralysis because both upper and lower motor neurons waste away.
There is no clear explanation as to why these motor neurons selectively degenerate. Several clues helped build the ‘dying-back hypothesis’, which postulates that ALS causes distal axons to lose their function and retract. It would explain why the longest and most energy-demanding motor neurons are among the most vulnerable ones.
FUS and transport defects
Genetic forms of ALS are rare, but can provide important insights into the disease mechanisms. One of the four major genes mutated in familial forms of ALS is FUS.
In collaboration with the Verfaillie lab at KU Leuven, the team of Prof. Ludo Van Den Bosch (VIB-KU Leuven) generated induced pluripotent stem cells from ALS patients with different FUS mutations. In this way, they could generate a new human neuronal model for the disease. Motor neurons derived from these stem cells showed typical cytoplasmic FUS mislocalization and hypoexcitability, but also progressive axonal transport defects of different cargoes, a pathological feature never observed before in these cells.
Dr. Wenting Guo, one of the main researchers involved in the study, explains: “Distal axonal sites are highly dependent on the supply of energy-producing organelles and other cargo’s from the cell nucleus, so the implication of axonal transport in ALS is not surprising. It is an important step that we can reproduce this feature of the disease in cultured human motor neurons.”
Axonal transport problems of mitochondria were previously described in models of mutant SOD1, which is also linked to familial ALS. In the case of SOD1, the transport defects were attributed to morphological changes in the mitochondria, but FUS mutations do not lead to gross mitochondrial damage. Wenting Guo: “Thanks to the expertise of our electron microscopy platform, we could demonstrate that mitochondria in FUS mutant neurons look healthy.”
HDAC6 to the rescue
CRISPR/Cas9-mediated genetic correction of the FUS mutation rescues the axonal transport defects, underscoring the specificity of the pathology. However, more interestingly, pharmacological inhibition or genetic silencing of HDAC6 also restores the axonal transport defects.
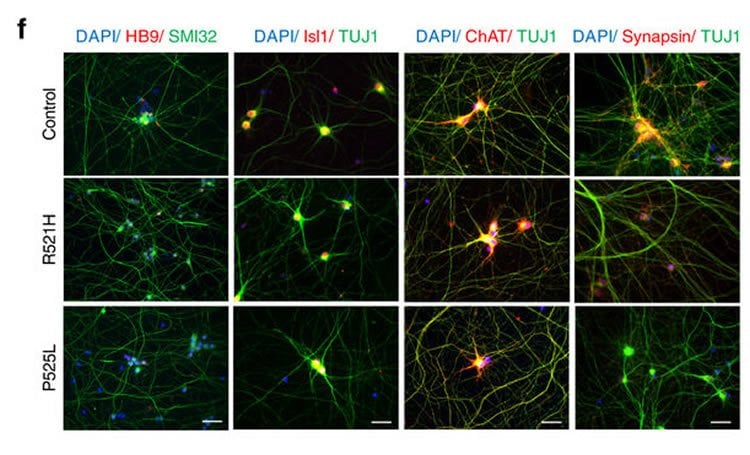
Van Den Bosch: “HDAC6 deacetylates the building blocks of the microtubules, the tracks used for axonal transport. When HDAC6 is inhibited, acetylation increases and axonal transport is improved. This may prevent axons from dying back.”
While he stresses that axonal transport dysfunction is only one aspect of the disease mechanism, Van Den Bosch is optimistic: “Axonal transport could play an important role in ALS pathology and HDAC6 inhibition could become a promising therapeutic approach, although stopping retraction alone might not be enough as a single therapeutic strategy.”
Source: Sooike Stoops – VIB Flanders.
Publisher: NeuroscienceNews.com.
Image Source: NeuroscienceNews.com images is credited to Van Den Bosch et al./Nature Communications.
Original Research: Full open access research for “HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients” by Wenting Guo, Maximilian Naujock, Laura Fumagalli, Tijs Vandoorne, Pieter Baatsen, Ruben Boon, Laura Ordovás, Abdulsamie Patel, Marc Welters, Thomas Vanwelden, Natasja Geens, Tine Tricot, Veronick Benoy, Jolien Steyaert, Cynthia Lefebvre-Omar, Werend Boesmans, Matthew Jarpe, Jared Sterneckert, Florian Wegner, Susanne Petri, Delphine Bohl, Pieter Vanden Berghe, Wim Robberecht, Philip Van Damme, Catherine Verfaillie & Ludo Van Den Bosch in Nature Communications. Published online October 11 2017 doi:10.1038/s41467-017-00911-y
[cbtabs][cbtab title=”MLA”]VIB Flanders. “Resolving Traffic Jams in Human ALS Motor Neurons.” NeuroscienceNews. NeuroscienceNews, 17 October 2017.
<https://neurosciencenews.com/als-motor-neurons-7755/>.[/cbtab][cbtab title=”APA”]VIB Flanders. (2017, October 17). Resolving Traffic Jams in Human ALS Motor Neurons. NeuroscienceNews. Retrieved October 17, 2017 from https://neurosciencenews.com/als-motor-neurons-7755/[/cbtab][cbtab title=”Chicago”]VIB Flanders. “Resolving Traffic Jams in Human ALS Motor Neurons.” https://neurosciencenews.com/als-motor-neurons-7755/ (accessed October 17, 2017).[/cbtab][/cbtabs]
Abstract
HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disorder due to selective loss of motor neurons (MNs). Mutations in the fused in sarcoma (FUS) gene can cause both juvenile and late onset ALS. We generated and characterized induced pluripotent stem cells (iPSCs) from ALS patients with different FUS mutations, as well as from healthy controls. Patient-derived MNs show typical cytoplasmic FUS pathology, hypoexcitability, as well as progressive axonal transport defects. Axonal transport defects are rescued by CRISPR/Cas9-mediated genetic correction of the FUS mutation in patient-derived iPSCs. Moreover, these defects are reproduced by expressing mutant FUS in human embryonic stem cells (hESCs), whereas knockdown of endogenous FUS has no effect, confirming that these pathological changes are mutant FUS dependent. Pharmacological inhibition as well as genetic silencing of histone deacetylase 6 (HDAC6) increase α-tubulin acetylation, endoplasmic reticulum (ER)–mitochondrial overlay, and restore the axonal transport defects in patient-derived MNs.
“HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients” by Wenting Guo, Maximilian Naujock, Laura Fumagalli, Tijs Vandoorne, Pieter Baatsen, Ruben Boon, Laura Ordovás, Abdulsamie Patel, Marc Welters, Thomas Vanwelden, Natasja Geens, Tine Tricot, Veronick Benoy, Jolien Steyaert, Cynthia Lefebvre-Omar, Werend Boesmans, Matthew Jarpe, Jared Sterneckert, Florian Wegner, Susanne Petri, Delphine Bohl, Pieter Vanden Berghe, Wim Robberecht, Philip Van Damme, Catherine Verfaillie & Ludo Van Den Bosch in Nature Communications. Published online October 11 2017 doi:10.1038/s41467-017-00911-y