

It’s the most common cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), but until now scientists weren’t sure how a specific gene caused these devastating diseases. Now researchers from the University of Toronto are one step closer to solving this incredibly complex puzzle, offering hope for treatment.
“Researchers knew that mutations in a specific gene caused 40 per cent of inherited cases of ALS, but there are few studies of the normal function of this gene,” said Janice Robertson, professor in the Faculty of Medicine’s Department of Laboratory Medicine and Pathobiology. “Other scientists have focused on how the gene’s mutation causes disease. We’ve developed the first antibodies to track what this gene does in both a normal and diseased cell.”
Using these antibodies, Robertson and her team tracked proteins from the key gene, C9orf72. They discovered that a specific protein from this gene might help transport other essential proteins in and out of a motor neuron cell’s nucleus, the command centre of the cell. Their findings were recently published in the Annals of Neurology.
“We saw that a protein from C9orf72 normally surrounds the nucleus of a motor neuron cell,” said Robertson, who is also a scientist at U of T’s Tanz Centre for Research in Neurodegenerative Diseases. “But in ALS or FTD, this protein moves to the outer membrane of the cell. When this protein is misplaced, it can’t help other proteins move in and out of the cell’s nucleus and the cell dies.”
This transportation normally depends on a cascade of events that acts like a relay race. DNA, the cell’s instructions, is converted into RNA which then makes proteins for the cell that allow it to carry out its functions. Just like a relay, when a protein from C9orf72 disappears from surrounding the nucleus, it can no longer help transport other essential proteins that keep the cell alive.
Next, the researchers want to understand how C9orf72 is involved in transporting these proteins and potentially target the pathway with drugs.
“This pathway is easily targeted with drugs that already exist,” said Dr. Shangxi Xiao, first author of the publication. “If we can restore function to this pathway and make proteins go back to the nucleus, we could develop treatment options.”
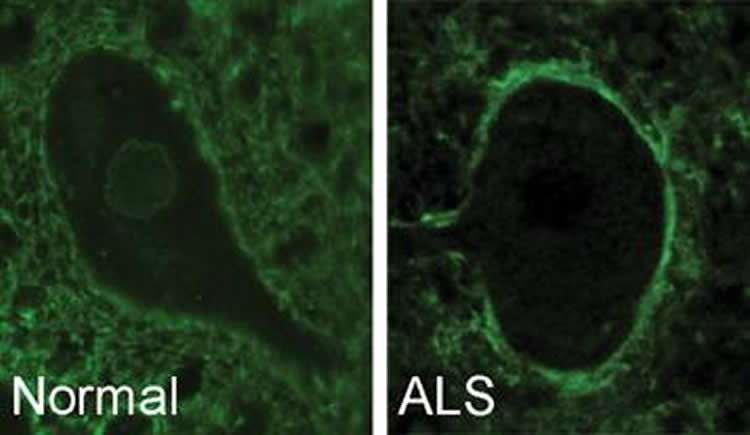
Robertson’s remarkable findings provide hope for treating ALS and FTD. ALS is a neuromuscular disease that begins as a mild muscle weakness and progresses to complete paralysis. Frontotemporal dementia is closely linked to ALS and leads to memory loss, behavior changes and difficulties with movement and speech.
“James Hunter is one of our financial supporters, and he’s completely paralyzed,” said Robertson. “He uses his eyes to communicate through a computer program, but now his eyes have almost stopped moving — this is ALS.”
Robertson believes the impact of this and others’ research will revolutionize treatment for ALS and FTD. “Recently, there have been four important publications, all honing in on this specific pathway. We’re getting closer to finding treatment options,” she said. “We’d like to thank those involved in the James Hunter and Family ALS Initiative and the families who have donated tissues for research. Ultimately, our goal is to find a treatment for patients who desperately need more options.”
Source: Liam Mitchell – University of Toronto
Image Source: The image is credited to University of Toronto
Original Research: Abstract for “Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis” by Shangxi Xiao, Laura MacNair B.Sc. (Hons), Philip McGoldrick PhD, Paul M. McKeever B.Sc. (Hons), Jesse R. McLean PhD, Ming Zhang PhD, Julia Keith MD, Lorne Zinman MD, Ekaterina Rogaeva PhD and Janice Robertson PhD in Annals of Neurology. Published online August 29 2015 doi:10.1002/ana.24469
Abstract
Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis
Objective
A noncoding hexanucleotide repeat expansion in C9orf72 is the most common cause of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). It has been reported that the repeat expansion causes a downregulation of C9orf72 transcripts, suggesting that haploinsufficiency may contribute to disease pathogenesis. Two protein isoforms are generated from three alternatively spliced transcripts of C9orf72; a long form (C9-L) and a short form (C9-S), and their function(s) are largely unknown owing to lack of specific antibodies.
Methods
To investigate C9orf72 protein properties, we developed novel antibodies that recognize either C9-L or C9-S. Multiple techniques, including Western blot, immunohistochemistry, and coimmunoprecipitation, were used to determine the expression levels and subcellular localizations of C9-L and C9-S.
Results
Investigation of expression of C9-L and C9-S demonstrated distinct biochemical profiles, region-specific changes, and distinct subcellular localizations in ALS tissues. In particular, C9-L antibody exhibited a diffuse cytoplasmic staining in neurons and labeled large speckles in cerebellar Purkinje cells. In contrast, C9-S antibody gave very specific labeling of the nuclear membrane in healthy neurons, with apparent relocalization to the plasma membrane of diseased motor neurons in ALS. Coimmunoprecipitation experiments revealed an interaction of the C9-isoforms with both Importin β1 and Ran-GTPase, components of the nuclear pore complex.
Interpretation
Using these antibodies, we have shown that C9orf72 may be involved in nucleocytoplasmic shuttling and this may have relevance to pathophysiology of ALS/FTLD. Our antibodies have provided improved detection of C9orf72 protein isoforms, which will help elucidate its physiological function and role in ALS/FTLD.
“Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis” by Shangxi Xiao, Laura MacNair B.Sc. (Hons), Philip McGoldrick PhD, Paul M. McKeever B.Sc. (Hons), Jesse R. McLean PhD, Ming Zhang PhD, Julia Keith MD, Lorne Zinman MD, Ekaterina Rogaeva PhD and Janice Robertson PhD in Annals of Neurology. Published online August 29 2015 doi:10.1002/ana.24469




