

Summary: Studying what the LRRK2 gene does in macrophages infected with the bacterium that causes tuberculosis, researchers believe they may have identified a cause of Parkinson’s disease.
Source: The Francis Crick Institute.
The mechanism our immune cells use to clear bacterial infections like tuberculosis (TB) might also be implicated in Parkinson’s disease, according to a new collaborative study led by the Francis Crick Institute, Newcastle University and GSK.
The findings, which will be published in The EMBO Journal, provide a possible explanation of the cause of Parkinson’s disease and suggest that drugs designed to treat Parkinson’s might work for TB too.
Parkinson’s protein
The most common genetic mutation in Parkinson’s disease patients is in a gene called LRRK2, which makes the LRRK2 protein overactive.
Drugs that block LRRK2 are a promising new treatment for Parkinson’s, with many pharmaceutical companies developing drugs to target LRRK2 and clinical trials underway. But how overactive LRRK2 causes Parkinson’s and why LRRK2 blockers work was a mystery.
The biological causes of Parkinson’s remain largely unknown, making it more difficult to develop and improve treatments. Discovering a mechanism that causes Parkinson’s and how drugs affect it could significantly advance efforts to improve treatments.
Insights from TB
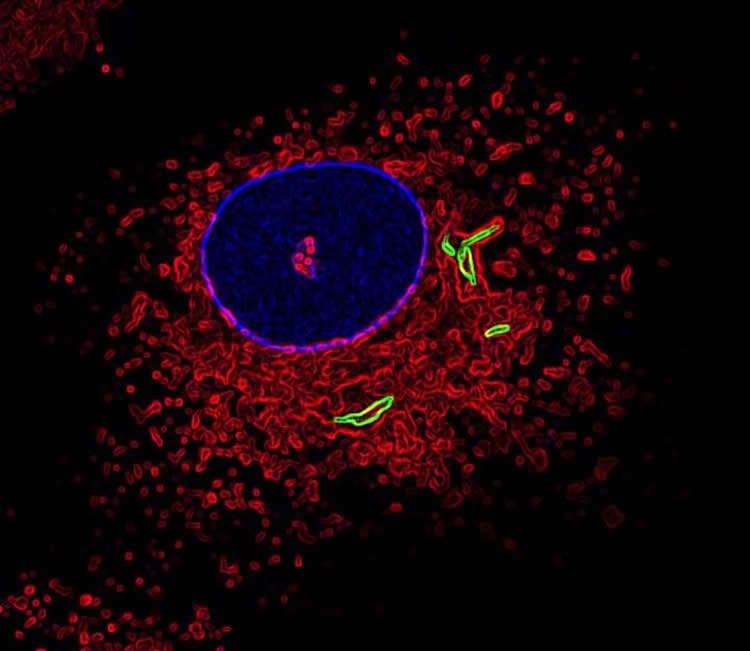
By studying what LRRK2 does in immune cells called macrophages that are infected with Mycobacterium tuberculosis (Mtb) – the bacterium that causes TB – researchers believe they have uncovered a potential cause of Parkinson’s.
Macrophages recognise and engulf Mtb securing it within tight-fitting internal compartments called phagosomes. Another part of the cell called the lysosome then fuses with the phagosome to destroy the bacterium inside.
Using a combination of experimental approaches, Crick and GSK researchers, in collaboration with proteomics specialist Matthias Trost from Newcastle University, found that LRRK2 prevents phagosomes from fusing with lysosomes in both human and mouse macrophages, making them less efficient at clearing bacteria. Deleting the LRRK2 gene or treating the cells with an LRRK2 blocker significantly reduced levels of Mtb.
These findings in cells were supported by experiments in mice. When the researchers deleted the gene for LRRK2 in mice, they found that they exhibited an enhanced early immune response to TB infection, and had significantly lower levels of Mtb in their lungs than control mice up to two weeks after infection.
“We think that this mechanism might also be at play in Parkinson’s disease, where abnormal masses of protein called ‘Lewy bodies’ build up in neurons in the brain and cause damage,” said Susanne Herbst, joint first author of the paper and post-doctoral fellow at the Crick.
The team suspect that LRRK2 might be preventing immune cells in the brain from degrading cell debris properly, leading to a build-up of protein in neurons that disrupts their function.
Susanne added: “By studying TB, we have found a possible explanation for why LRRK2 mutations are a genetic risk factor for Parkinson’s disease. It’s exciting when different fields of research connect up in unexpected ways like this!”
Co-author Patrick Lewis, Associate Professor in Cellular and Molecular Neuroscience at the University of Reading, said: “The dogma in the Parkinson’s field has been to focus almost exclusively on what is happening to neurons in the brain to make them degenerate. But over the last few years, there has been a growing appreciation of the integral role of other cells in the brain and particularly the immune system in keeping neurons healthy. This study reinforces why we should think more broadly about the events that cause neurodegeneration, and that some of the answers to Parkinson’s disease might come from immunology.”
New TB treatments
The findings also suggest that LRRK2 inhibitors could be a powerful new way of combating TB, which kills 1.67 million people every year.
“Drug-resistant TB is a serious emerging problem, and boosting the body’s own immune defence against TB is an important step in the battle against antibiotic resistant strains,” said Max Gutierrez, Group Leader at the Crick and senior author of the paper.
“LRRK2 inhibiting drugs are already being developed to treat Parkinson’s disease and we’re trying to see if we can repurpose them as a potential new TB therapy. This should be relatively straightforward because TB infects the lungs, so the LRRK2 inhibitors wouldn’t need to cross the blood-brain barrier like they do in Parkinson’s disease.”
Source: Greta Keenan – The Francis Crick Institute
Publisher: Organized by NeuroscienceNews.com.
Image Source: NeuroscienceNews.com image is credited to Susanne Herbst, Francis Crick Institute.
Original Research: Open access research for “LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophagesa” by Anetta Härtlova, Susanne Herbst, Julien Peltier, Angela Rodgers, Orsolya Bilkei‐Gorzo, Antony Fearns, Brian D Dill, Heyne Lee, Rowan Flynn, View ORCID ProfileSally A Cowley, Paul Davies, Patrick A Lewis, Ian G Ganley, Jennifer Martinez, Dario R Alessi, Alastair D Reith, Matthias Trost, and Maximiliano G Gutierrez in EMBO Journal. Published May 22 2018
doi:10.15252/embj.201798694
[cbtabs][cbtab title=”MLA”]The Francis Crick Institute “Link Between Tuberculosis and Parkinson’s Disease Discovered.” NeuroscienceNews. NeuroscienceNews, 22 May 2018.
<https://neurosciencenews.com/parkinsons-tb-9111/>.[/cbtab][cbtab title=”APA”]The Francis Crick Institute (2018, May 22). Link Between Tuberculosis and Parkinson’s Disease Discovered. NeuroscienceNews. Retrieved May 22, 2018 from https://neurosciencenews.com/parkinsons-tb-9111/[/cbtab][cbtab title=”Chicago”]The Francis Crick Institute “Link Between Tuberculosis and Parkinson’s Disease Discovered.” https://neurosciencenews.com/parkinsons-tb-9111/ (accessed May 22, 2018).[/cbtab][/cbtabs]
Abstract
LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages
Mutations in the leucine‐rich repeat kinase 2 (LRRK2) are associated with Parkinson’s disease, chronic inflammation and mycobacterial infections. Although there is evidence supporting the idea that LRRK2 has an immune function, the cellular function of this kinase is still largely unknown. By using genetic, pharmacological and proteomics approaches, we show that LRRK2 kinase activity negatively regulates phagosome maturation via the recruitment of the Class III phosphatidylinositol‐3 kinase complex and Rubicon to the phagosome in macrophages. Moreover, inhibition of LRRK2 kinase activity in mouse and human macrophages enhanced Mycobacterium tuberculosis phagosome maturation and mycobacterial control independently of autophagy. In vivo, LRRK2 deficiency in mice resulted in a significant decrease in M. tuberculosis burdens early during the infection. Collectively, our findings provide a molecular mechanism explaining genetic evidence linking LRRK2 to mycobacterial diseases and establish an LRRK2‐dependent cellular pathway that controls M. tuberculosis replication by regulating phagosome maturation.




