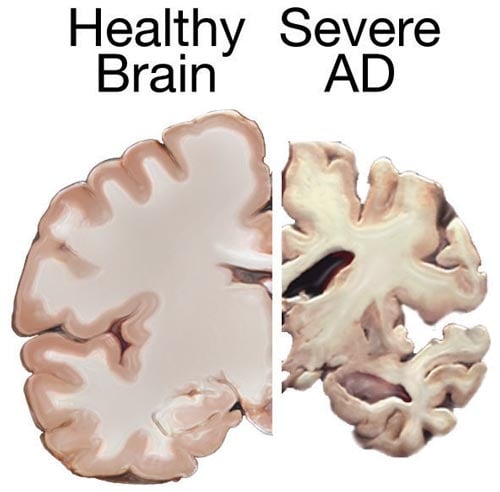
Finding how impaired activity of ADAM10 enzyme increases neurodegeneration suggests therapeutic applications.
Massachusetts General Hospital (MGH) researchers have identified and validated two rare gene mutations that appear to cause the common form of Alzheimer’s disease (AD) that strikes after the age of 60. The two mutations occur in a gene called ADAM10 – coding for an enzyme involved in processing the amyloid precursor protein – which now becomes the second pathologically-confirmed gene for late-onset AD and the fifth AD gene overall.
In their report, which will appear in the October 16 issue of Neuron and has been released online, the investigators from the Mass General Institute for Neurodegenerative Disease (MGH-MIND) describe how the two mutations in ADAM10 increase generation and accumulation of the toxic amyloid beta (A-beta) protein in the brains of a mouse model of AD. The mutations also reduce generation of new neural cells in hippocampus, a part of the brain essential to learning and memory.

“This is the first report to document, in animal models, new pathogenic gene mutations for AD since the reports of the original four genes in the 1990s,” says Rudolph Tanzi, PhD, director of the Genetics and Aging Research Unit at MGH-MIND and senior author of the Neuron paper. “What we found regarding the many effects of these two rare mutations in ADAM10 strongly suggests that diminished activity of this enzyme can cause AD, and these findings support ADAM10 as a promising therapeutic target for both treatment and prevention.”
The process leading to the generation of A-beta – which accumulates in characteristic plaques in the brains of AD patients – begins when the amyloid precursor protein (APP) is cut into smaller proteins by enzymes called secretases. A-beta results if APP is first cut into two segments by an enzyme called beta-secretase, and one of those segments is further cut by a gamma-secretase enzyme to release the toxic A-beta fragment. However, processing of APP by an alpha-secretase enzyme – one of which is ADAM10 – cuts right through the A-beta region in APP. So instead of generating the toxic A-beta fragment, cleavage with alpha-secretase produces a protein fragment that has been reported to protect and stimulate the generation of neurons in brain.
An earlier study by Tanzi’s team reported finding that either of two mutations in ADAM10 increased the risk of AD in seven families with the late-onset form of the disease. Since ADAM10 was already known to be important to alpha-secretase processing of APP, along with having a role in early brain development, the researchers set out to investigate how the observed mutations might lead to the pattern of neurodegeneration characteristic of AD.
Experiments using several strains of transgenic mice – including lines that express both one of the ADAM10 mutations and an APP mutation that leads to AD-like pathology – revealed the following:
- AD-associated mutations in ADAM10 reduced the release from neurons in the animals’ brains of the beneficial protein produced by alpha-secretase processing of APP,
- Reduced ADAM10 activity caused by the mutations increased the generation of A-beta and its accumulation in plaques, along with producing other AD-associated neurodegenerative signs,
- Reduced ADAM10 activity also impaired the generation of new neurons in the hippocampus, one of the areas of the brain most vulnerable to neurodegeneration in AD,
- The AD-associated mutations produce these effects by impairing the correct folding of ADAM10 and interfering with its normal functions.
Jaehong Suh, PhD, of the MGH-MIND Genetics and Aging Research Unit, lead author of the Neuron article, says, “Our current study shows that reducing ADAM10 activity by these AD-associated mutations delivers a ‘one-two punch’ to the brain – one, decreasing neuroprotective alpha-secretase cleavage products and two, increasing neurotoxic A-beta protein accumulation. Thus, we believe that increasing ADAM10 activity might help to alleviate both genetic and environmental AD risk factors that increase the toxic beta-secretase processing of APP. We’re planning to develop optimal ways to increase ADAM10 activity in brain and to further investigate the molecular structure and regulatory mechanism of the ADAM10 enzyme.” Suh is an instructor in Neurology, and Tanzi is the Joseph P. and Rose F. Kennedy Professor of Neurology at Harvard Medical School.
Notes about this neurogenetics and Alzheimer’s disease research
Additional co-authors of the report are Se Hoon Choi, PhD, Donna Romano, Moira Gannon, Andrea Lesinski, and Doo Yeon Kim, PhD, all of the MGH-MIND Genetics and Aging Research Unit. The study was supported by the Cure Alzheimer’s Fund and grants from the National Institute on Aging, the National Institute for Mental Health and the American Health Assistance Foundation.
Contact: Mike Morrison – Massachusetts General Hospital
Source: Massachusetts General Hospital press release
Image Source: The Alzheimer’s brain slice image is credited to the NIH and is in the public domain.
Original Research: Abstract for “ADAM10 Missense Mutations Potentiate β-Amyloid Accumulation by Impairing Prodomain Chaperone Function” by Jaehong Suh, Se Hoon Choi, Donna M. Romano, Moira A. Gannon, Andrea N. Lesinski, Doo Yeon Kim, Rudolph E. Tanzi in Neuron. Published online September 19 2013 doi:10.1016/j.neuron.2013.08.035