Summary: Researchers report the injury pathway is triggered when Unc-104 is mutated. This mutation leads synapses to become defective.
Source: University of Michigan.
An injury pathway in the neurons of fruit flies may cause the loss of synapses in diseases such as Alzheimer’s and ALS, according to University of Michigan researchers.
This pathway, called DLK, has received recent attention as a candidate drug target because it contributes to the deterioration of damaged neurons. The new findings further expand that interest by suggesting that inhibiting DLK may help neurons to maintain working synapses, which is more useful than simply preventing damaged neurons from dying.
The pathway, studied here in fruit flies, is similar to the pathway in the neurons of mammals and humans. The U-M team, which includes former doctoral student Jiaxing Li and Catherine Collins, associate professor of molecular, cellular and developmental biology, found a new relationship between the injury pathway and a protein in neurons called kinesin—specifically, a kinesin called Unc-104.
When neurons communicate with each other, they extend a projection called an axon toward each other and form sites for information exchange called synapses. Transmitting information between synapses requires many protein molecules, which are manufactured within the neuron cell body and ferried over long distances within axons by kinesins such as Unc-104. The Unc-104 kinesin seems to carry many of the proteins needed for the neuron to release neurotransmitters for information exchange.
The U-M researchers found that the injury pathway is triggered when Unc-104 is damaged or mutated. This builds upon previous knowledge that the pathway is required for the key responses neurons make when damaged, including initiating the neuron to repair itself or die, depending on the context.
When Unc-104 is damaged or mutated, synapses become defective. Previously, researchers assumed that this is because of a failure to transport synaptic proteins to synapse sites. However, in their study, Li, Collins and their team found that turning off the neuronal injury pathway can restore the function of these mutant synapses. Their results are published in the journal eLife.
To study the role of the kinesin and the injury pathway in the malfunction of synapses, the researchers imaged the synapses of dissected fruit fly larvae using a confocal microscope. Working in the neighboring lab of U-M professor Richard Hume, Li also used electrophysiology to measure how well the synapses were firing. The team found that when they shut down the injury pathway, the function of that synapse was restored.
“That was really striking,” Collins said. “It told us that the axon injury pathway was causing these major problems in synapses.”
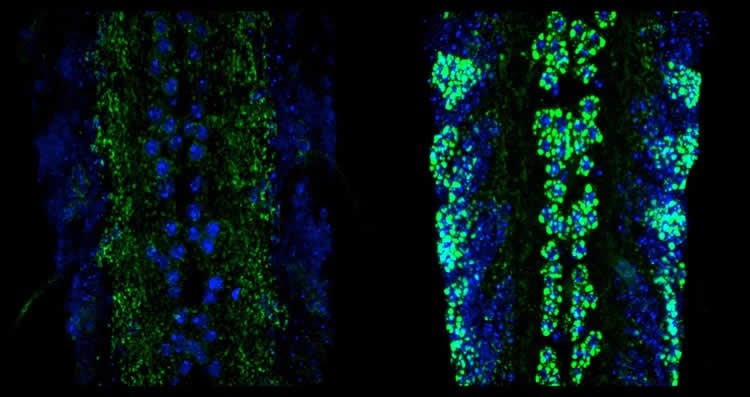
The team found that the pathway becomes activated when the Unc-104 kinesin is impaired, and that once activated, the pathway shuts down the formation of many of the synaptic proteins which are normally transported in axons.
“In the fruit fly system, we know the neuroanatomy really well. We can actually see how impairment of kinesin is affecting things in cell bodies, synapses and neurons,” Collins said. “When we turned down the injury pathway in the kinesin mutants, we could see a huge mass of synaptic proteins accumulating in the cell body. That led us to the idea that the injury pathway turns down the levels of many synaptic proteins, leading to synapse malfunction.”
The findings imply that activation of the injury pathway has negative consequences for synapses. Complementing this work, recent findings published in Science Translational Medicine suggest that the injury pathway may be activated in patients with neurodegenerative diseases ALS and Alzheimer’s disease.
Restricting the pathway can also delay symptoms of these diseases in mouse models. This draws attention to the pathway as a potential therapeutic target.
But this could also hinder beneficial functions of the injury pathway, Collins says.
“When the injury pathway was knocked down in flies, the massive accumulations of proteins in cell bodies suggested to us that it functions as a stress response mechanism, to prevent unwanted build-up of proteins when axonal transport is impaired,” she said.
Next, the researchers want to understand why the injury pathway appears specifically tuned to Unc-104 for its activation, and how the pathway reduces synaptic proteins.
Source: Morgan Sherburne – University of Michigan
Image Source: NeuroscienceNews.com image is credited to Jiaxing Li.
Original Research: Full open access research for “Restraint of presynaptic protein levels by Wnd/DLK signaling mediates synaptic defects associated with the kinesin-3 motor Unc-104” by Jiaxing Li, Yao V Zhang, Elham Asghari Adib, Doychin T Stanchev, Xin Xiong, Susan Klinedinst, Pushpanjali Soppina, Thomas Robert Jahn, Richard I Hume, Tobias M Rasse, and Catherine A Collins in eLife. Published online September 19 2017 doi:10.7554/eLife.24271
[cbtabs][cbtab title=”MLA”]University of Michigan “Pathway in Neurons May Contribute to Neurodegenerative Diseases.” NeuroscienceNews. NeuroscienceNews, 4 October 2017.
<https://neurosciencenews.com/neurodegeneration-neuron-pathway-7656/>.[/cbtab][cbtab title=”APA”]University of Michigan (2017, October 4). Pathway in Neurons May Contribute to Neurodegenerative Diseases. NeuroscienceNews. Retrieved October 4, 2017 from https://neurosciencenews.com/neurodegeneration-neuron-pathway-7656/[/cbtab][cbtab title=”Chicago”]University of Michigan “Pathway in Neurons May Contribute to Neurodegenerative Diseases.” https://neurosciencenews.com/neurodegeneration-neuron-pathway-7656/ (accessed October 4, 2017).[/cbtab][/cbtabs]
Abstract
Restraint of presynaptic protein levels by Wnd/DLK signaling mediates synaptic defects associated with the kinesin-3 motor Unc-104
The kinesin-3 family member Unc-104/KIF1A is required for axonal transport of many presynaptic components to synapses, and mutation of this gene results in synaptic dysfunction in mice, flies and worms. Our studies at the Drosophila neuromuscular junction indicate that many synaptic defects in unc-104-null mutants are mediated independently of Unc-104’s transport function, via the Wallenda (Wnd)/DLK MAP kinase axonal damage signaling pathway. Wnd signaling becomes activated when Unc-104’s function is disrupted, and leads to impairment of synaptic structure and function by restraining the expression level of active zone (AZ) and synaptic vesicle (SV) components. This action concomitantly suppresses the buildup of synaptic proteins in neuronal cell bodies, hence may play an adaptive role to stresses that impair axonal transport. Wnd signaling also becomes activated when pre-synaptic proteins are over-expressed, suggesting the existence of a feedback circuit to match synaptic protein levels to the transport capacity of the axon.
“Restraint of presynaptic protein levels by Wnd/DLK signaling mediates synaptic defects associated with the kinesin-3 motor Unc-104” by Jiaxing Li, Yao V Zhang, Elham Asghari Adib, Doychin T Stanchev, Xin Xiong, Susan Klinedinst, Pushpanjali Soppina, Thomas Robert Jahn, Richard I Hume, Tobias M Rasse, and Catherine A Collins in eLife. Published online September 19 2017 doi:10.7554/eLife.24271