UC San Francisco scientists working in the lab used a chemical found in an anti-wrinkle cream to prevent the death of nerve cells damaged by mutations that cause an inherited form of Parkinson’s disease. A similar approach might ward off cell death in the brains of people afflicted with Parkinson’s disease, the team suggested in a study reported online in the journal Cell on August 15.
The achievement marks a pharmacologic milestone as the first highly specific targeting of a member of an important class of enzymes called kinases to increase rather than to inhibit their activity, according to UCSF chemist Kevan Shokat, PhD, the senior scientist on the study. The research raises hope that similar pharmaceutical strategies might be used for combatting other diseases, including diabetes and cancer, he said.

Mutations that cause malfunction of the targeted enzyme, PINK1, are directly responsible for some cases of early-onset Parkinson’s disease. Loss of PINK1 activity is harmful to the cell’s power plants, called mitochondria, best known for converting food energy into another form of chemical energy used by cells, the molecule ATP.
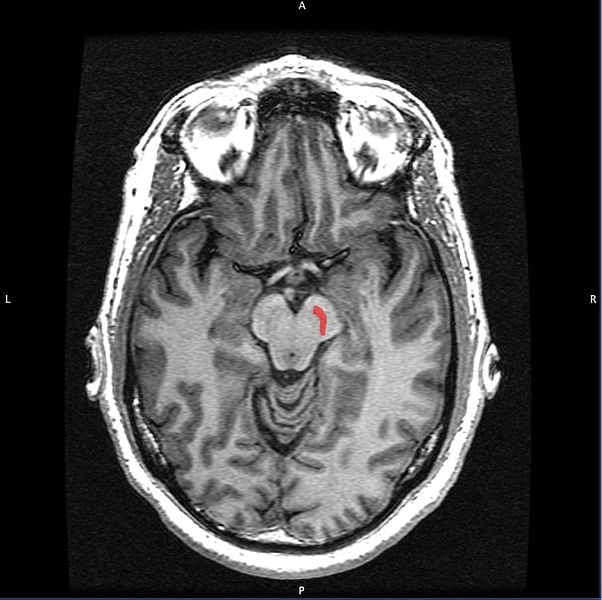
In Parkinson’s disease, poorly performing mitochondria have been associated with the death of dopamine-producing nerve cells in a region of the brain called the substantia nigra, which plays a major role in control of movement. Loss of these cells is a hallmark of Parkinson’s disease and the cause of prominent symptoms including rigidity and tremor.
A UCSF team led by Shokat, a Howard Hughes Medical Institute Investigator, used the chemical, called kinetin, to increase mutant PINK1 enzyme activity in nerve cells to near normal levels.
“In light of the fact that mutations in PINK1 produce Parkinson’s disease in humans, the finding that kinetin can speed mutated PINK1 activity to near normal levels raises the possibility that kinetin may be used to treat these patients,” Shokat said.
The researchers also found that, in nerve cells with normal PINK1, kinetin boosted enzyme activity beyond typical levels. This finding may be relevant for the most common forms of Parkinson’s disease, in which PINK1 is not mutated, because a previous study showed that similar overactivity of PINK1 can slow the development of abnormal movement in a fruit-fly model of Parkinson’s disease caused by another defect. This defect is elevated production of the protein alpha-synuclein, also a cause of some inherited cases of Parkinson’s disease.
The demonstration in the new study that PINK1 can be boosted in human nerve cells that lack PINK1 mutations therefore suggests that kinetin might also have therapeutic potential in common cases of Parkinson’s disease in which PINK1 is not mutated, Shokat said.
Parkinson’s disease is the second most common neurodegenerative disease after Alzheimer’s disease, and the 14th leading cause of death in the United States, according to the U.S Centers for Disease Control and Prevention. Current treatments primarily aim to boost availability of dopamine to brain regions where dopamine-producing nerve cells have been lost.
Although many drugs that inhibit the activity of kinases have been developed over the past decade, including 15 currently approved to treat cancer, Shokat said none has yet been marketed to directly boost activity of a kinase.
The breakthrough in revving up PINK1 activity pharmacologically stemmed from Shokat’s unconventional approach. He targeted the enzyme’s “substrate,” a molecule that binds to an enzyme and undergoes a quick chemical transformation as a result. PINK1 uses ATP as a substrate, and the chemical reaction helps PINK1 in turn drive the activation of another enzyme, called Parkin.
Both of these enzymes are among a small number that previously have been strongly linked to Parkinson’s disease. PINK1 and Parkin act together to monitor the health of mitochondria, and help trigger repair or disposal of damaged mitochondria within the cells, thereby promoting cell survival.
“Therapeutic approaches for enhancing the activity of PINK1 had not been considered, because scientists had not conceived of the idea of developing a new substrate for the enzyme,” Shokat said.
“We found that a small molecule, called KTP, speeds chemical reactions catalyzed by PINK1 better than ATP, the natural substrate. That kind of better-than-natural response is essentially unheard of.”
KTP is too big to fit into other kinases, Shokat said, but PINK1 has a larger ATP “pocket” to hold KTP.
After discovering the potential of KTP, the researchers then determined that kinetin is converted to KTP within cells. Experimentally, kinetin, which can cross blood vessels to get into the brain, has been given by mouth to treat a rare, genetic, neurological disease called familial dysautonomia.
Notes about this Parkinson’s disease research
Other researchers on the UCSF study include graduate student Nicholas Hertz, PhD; post-doctoral fellows Martin Sos, PhD and Amandine Berthet, PhD; UCSF faculty members Ken Nakamura, MD, PhD from the Gladstone Institute, and Kurt Thorn, PhD, and Al Burlingame, PhD.
The research was funded by the National Institutes for Health and by the Michael J. Fox Foundation.
Hertz and Shokat are inventors on a patent application related to kinetin and PINK1. UCSF has licensed the patent application to Mitokinin LLC, and Hertz and Shokat are cofounders and members of the company.
Contact: Jeffrey Norris – UCSF
Source: UCSF press release
Image Source: The substantia nigra brain scan image is credited to Geoff B Hall and is licensed as public domain.
Original Research: Abstract for “A Neo-Substrate that Amplifies Catalytic Activity of Parkinson’s-Disease-Related Kinase PINK1” by Nicholas T. Hertz, Amandine Berthet, Martin L. Sos, Kurt S. Thorn, Al L. Burlingame, Ken Nakamura and Kevan M. Shokat in Cell. Published online August 15 2013 doi:10.1016/j.cell.2013.07.030