

Two proteins that share the ability to help cells deal with their trash appear to need each other to do their jobs and when they don’t connect, it appears to contribute to development of Parkinson’s disease, scientists report.
Much like a community’s network for garbage handling, cells also have garbage sites called lysosomes, where proteins, which are functioning badly because of age or other reasons, go for degradation and potential recycling, said Dr. Wen-Cheng Xiong, developmental neurobiologist and Weiss Research Professor at the Medical College of Georgia at Georgia Regents University.
Inside lysosomes, other proteins, called proteases, help cut up proteins that can no longer do their job and enable salvaging of things like precious amino acids. It’s a normal cell degradation process called autophagy that actually helps cells survive and is particularly important in cells such as neurons, which regenerate extremely slowly, said Xiong, corresponding author of the study in The Journal of Neuroscience.
Key to the process – and as scientists have shown, to each other – are two more proteins, VPS35 and Lamp2a. VPS35 is essential for retrieving membrane proteins vital to cell function. Levels naturally decrease with age, and mutations in the VPS35 gene have been found in patients with a rare form of Parkinson’s. VPS35 also is a critical part of a protein complex called a retromer, which has a major role in recycling inside cells. Lamp2a enables unfit proteins to be chewed up and degraded inside lysosomes.
If the two sound like a natural couple, scientists now have more evidence that they are. They have shown that without VPS35 to retrieve Lamp2a from the trash site for reuse, Lamp2a, or lysosomal-associated membrane protein 2, will be degraded and its vital function lost.
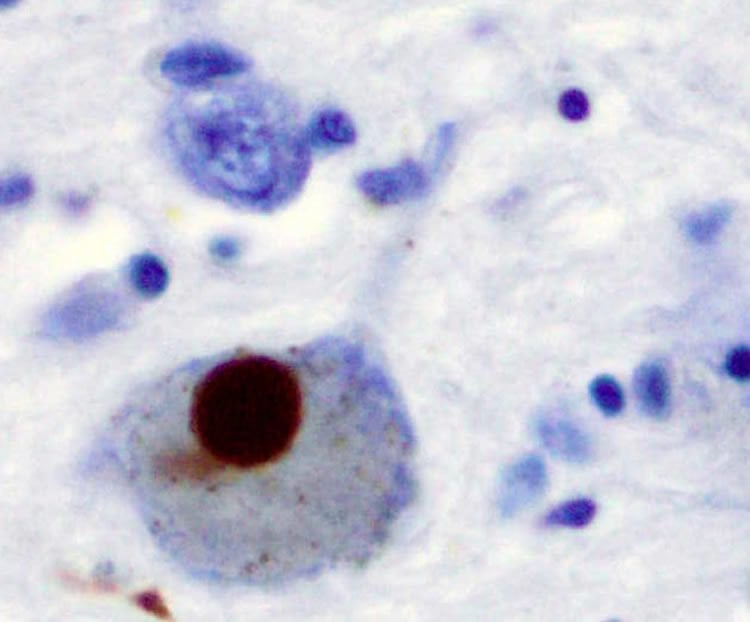
When the scientists generated VPS35-deficient mice, the mice exhibited Parkinson’s-like deficits, including impaired motor control. When they looked further, they found the lysosomes inside dopamine neurons, which are targets in Parkinson’s, didn’t function properly in the mice. In fact, without VPS35, the degradation of Lamp2a itself is accelerated. Consequently, yet another protein, alpha-synuclein, which is normally destroyed by Lamp2a, is increased. Alpha-synuclein is a major component of abnormal protein clumps, called Lewy bodies, found in the brains of patients with Parkinson’s.
“If alpha-synuclein is not degraded, it just accumulates. If VPS35 function is normal, we won’t see its accumulation,” Xiong said.
Conversely, when MCG scientists increased expression of Lamp2a in the dopamine neurons of the VPS35-deficient mice, alpha-synuclein levels were reduced, a finding that further supports the linkage of the three proteins in the essential ability of the neurons to deal with undesirables in their lysosomes.
Without lamp2a, dopamine neurons essentially start producing more garbage rather than eliminating it. Recycling of valuables such as amino acids basically stops, and alpha-synuclein is free to roam to other places in the cell or other brain regions where it can damage still viable proteins.
The bottom line is dopamine neurons are lost instead of preserved. Brain scans document the empty spaces where neurons used to be in patients with neurodegenerative diseases such as Parkinson’s and Alzheimer’s. One of the many problems with treatment of these diseases is that by the time the empty spaces and sometimes the associated symptoms are apparent, much damage has occurred, Xiong said.
Putting these pieces together provides several new, early targets for disease intervention. “Everything is linked,” Xiong said.
Dopamine is a brain chemical with many roles, including motor control, and patients with Parkinson’s have a loss of the neurons that secrete this neurotransmitter. At least in mice, VPS35 is naturally expressed in dopamine neurons in areas of the brain affected by Parkinson’s.
Xiong and her colleagues reported in 2011 that reduced expression of VPS35 enables activity of the dormant-in-healthy-adults protein BACE1 to increase along with accumulation of the brain plaque that is a hallmark of Alzheimer’s. Xiong said then that impaired VPS35 function likely also was a factor in Parkinson’s.
In a definite vicious circle, trash starts overwhelming the brain cell’s natural garbage disposal system. Proteins start getting misfolded and dysfunctional, potentially destructive proteins such as BACE1 and Lamp2a end up in the wrong place and get activated/inactivated, while good proteins get chopped up and/or bad proteins accumulate.
Parkinson’s disease is characterized by uncontrolled shaking, an unstable gait and cognitive loss. The research was funded by the National Institutes of Health and the Department of Veterans Affairs. Postdoctoral Fellow Dr. Fulei Tang is the study’s first author.
Source: Toni Baker – Georgia Regents University
Image Credit: The image is credited to Marvin 101 and is licensed CC BY-SA 3.0
Original Research: Abstract for “VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease” by Fu-Lei Tang, Joanna R. Erion, Yun Tian, Wei Liu, Dong-Min Yin, Jian Ye, Baisha Tang, Lin Mei, and Wen-Cheng Xiong in Journal of Neuroscience. Published online July 22 2015 doi:10.1523/JNEUROSCI.0042-15.2015
Abstract
VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease
Vacuolar protein sorting-35 (VPS35) is essential for endosome-to-Golgi retrieval of membrane proteins. Mutations in the VPS35 gene have been identified in patients with autosomal dominant PD. However, it remains poorly understood if and how VPS35 deficiency or mutation contributes to PD pathogenesis. Here we provide evidence that links VPS35 deficiency to PD-like neuropathology. VPS35 was expressed in mouse dopamine (DA) neurons in substantia nigra pars compacta (SNpc) and STR (striatum)—regions that are PD vulnerable. VPS35-deficient mice exhibited PD-relevant deficits including accumulation of α-synuclein in SNpc-DA neurons, loss of DA transmitter and DA neurons in SNpc and STR, and impairment of locomotor behavior. Further mechanical studies showed that VPS35-deficient DA neurons or DA neurons expressing PD-linked VPS35 mutant (D620N) had impaired endosome-to-Golgi retrieval of lysosome-associated membrane glycoprotein 2a (Lamp2a) and accelerated Lamp2a degradation. Expression of Lamp2a in VPS35-deficient DA neurons reduced α-synuclein, supporting the view for Lamp2a as a receptor of chaperone-mediated autophagy to be critical for α-synuclein degradation. These results suggest that VPS35 deficiency or mutation promotes PD pathogenesis and reveals a crucial pathway, VPS35-Lamp2a-α-synuclein, to prevent PD pathogenesis.
SIGNIFICANCE STATEMENT VPS35 is a key component of the retromer complex that is essential for endosome-to-Golgi retrieval of membrane proteins. Mutations in the VPS35 gene have been identified in patients with PD. However, if and how VPS35 deficiency or mutation contributes to PD pathogenesis remains unclear. We demonstrated that VPS35 deficiency or mutation (D620N) in mice leads to α-synuclein accumulation and aggregation in the substantia nigra, accompanied with DA neurodegeneration. VPS35-deficient DA neurons exhibit impaired endosome-to-Golgi retrieval of Lamp2a, which may contribute to the reduced α-synuclein degradation through chaperone-mediated autophagy. These results suggest that VPS35 deficiency or mutation promotes PD pathogenesis, and reveals a crucial pathway, VPS35-Lamp2a-α-synuclein, to prevent PD pathogenesis.
“VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease” by Fu-Lei Tang, Joanna R. Erion, Yun Tian, Wei Liu, Dong-Min Yin, Jian Ye, Baisha Tang, Lin Mei, and Wen-Cheng Xiong in Journal of Neuroscience. Published online July 22 2015 doi:10.1523/JNEUROSCI.0042-15.2015




