A discovery into the mechanisms which lead to degeneration and loss of communication among neuron cells – the cells controlling function in the brain and nervous system – could potentially lead to future therapies for neurodegenerative diseases including Parkinson’s and Alzheimer’s.
Scientists at The University of Nottingham have discovered that a small molecule called nicotinamide mononucleotide (NMN) causes a chain reaction of destruction within the neuron cell processes, called axons.
The research, published in the academic journal Cell Reports and conducted by researchers in the University’s School of Life Sciences, was led by Dr Laura Conforti and PhD student Andrea Loreto.
Neurons are extraordinary, highly compartmentalised cells that communicate and transmit electrical signals to other cells in the body through their axons, which are very long and slender projections making up to 99 per cent of the cell’s volume.
Their delicate shape and important function make axons very vulnerable and sensitive to early degeneration in a range of age-related neurodegenerative disorders including Parkinson’s disease, Huntington’s disease, Alzheimer’s disease, motor neuron disease and Multiple Sclerosis and in traumatic nerve injuries, preventing nerve cells from communicating with one another and with other cells and often causing symptoms.
The scientists have previously published research into NMN, which is known as a precursor to nicotinamide adenine dinucleotide (NAD), a coenzyme and ‘helper molecule’ present in all living cells that is essential for the production of the cell’s energy. This research showed that NMN initiates axon degeneration both in neurons in lab cultures and in zebrafish models of acute injury and neurotoxicity.
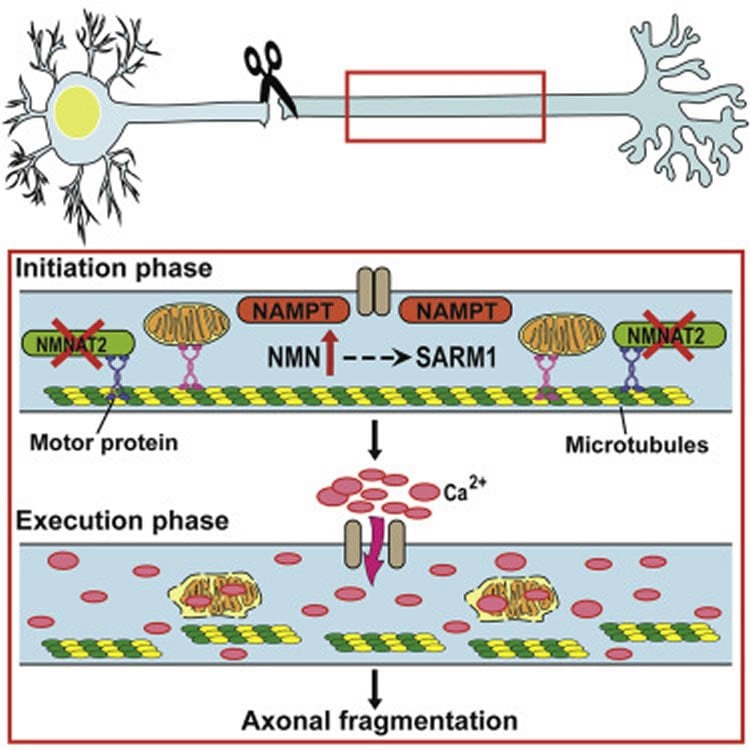
Building on the earlier work, the current study demonstrates that NMN in injured axons works together with SARM1, a protein involved in the innate immune system – the body’s first line of defence against potential pathogens – that plays a key role in axon degeneration, and kicks off a chain reaction leading to toxic levels of calcium and axon fragmentation.
The latest work again used cultured neurons and zebrafish models, the latter set up in collaboration with Dr Martin Gering, employing advanced microscopy techniques, with the assistance of the School of Life Sciences Imaging (SLIM) facility led by Experimental Chief Officer Tim Self.
Dr Laura Conforti said: “This study adds a new layer to our understanding about the connections between NAD metabolism and axon degeneration as it identifies a signalling role for NMN, a molecule previously known only as an NAD precursor.
“This discovery suggests that controlling the levels of NMN and the downstream signals it evokes may have an unexpected therapeutic potential in neurological disorders where axon degeneration is an underlying cause.
“Clinical applications of our research still require extensive further studies. However our findings identify important players in the chain of events leading to axon death and the steps that are the most promising targets for therapy.”
Funding: This study was supported by the National Institute of Health and the JPB foundation, with additional support from Stanford School of Medicine, Vincent and Stella Coates, and the Sheldon and Miriam Adelson Medical Research Foundation.
Source: Emma Thorne – University of Nottingham
Image Source: The image is credited to Conforti et al./Cell Reports
Original Research: Full open access research for “Wallerian Degeneration Is Executed by an NMN-SARM1-Dependent Late Ca2+ Influx but Only Modestly Influenced by Mitochondria” by Andrea Loreto, Michele Di Stefano, Martin Gering, and Laura Conforti in Cell Reports. Published online December 10 2015 doi:10.1016/j.celrep.2015.11.032
Abstract
Wallerian Degeneration Is Executed by an NMN-SARM1-Dependent Late Ca2+ Influx but Only Modestly Influenced by Mitochondria
Highlights
•NMN stimulates an intra-axonal Ca2+ rise shortly preceding axonal fragmentation
•NMN requires SARM1 to induce Ca2+ rise and axon degeneration
•The extracellular environment is the main source of NMN-induced axonal Ca2+ rise
•Mitochondrial dynamic changes are not causative in NMN-induced degeneration
Summary
Axon injury leads to rapid depletion of NAD-biosynthetic enzyme NMNAT2 and high levels of its substrate, NMN. We proposed a key role for NMN in Wallerian degeneration but downstream events and their relationship to other mediators remain unclear. Here, we show, in vitro and in vivo, that axotomy leads to a late increase in intra-axonal Ca2+, abolished by pharmacological or genetic reduction of NMN levels. NMN requires the pro-degenerative protein SARM1 to stimulate Ca2+ influx and axon degeneration. While inhibition of NMN synthesis and SARM1 deletion block Ca2+ rise and preserve axonal integrity, they fail to prevent early mitochondrial dynamic changes. Furthermore, depolarizing mitochondria does not alter the rate of Wallerian degeneration. These data reveal that NMN and SARM1 act in a common pathway culminating in intra-axonal Ca2+ increase and fragmentation and dissociate mitochondrial dysfunctions from this pathway, elucidating which steps may be most effective as targets for therapy.
“Wallerian Degeneration Is Executed by an NMN-SARM1-Dependent Late Ca2+ Influx but Only Modestly Influenced by Mitochondria” by Andrea Loreto, Michele Di Stefano, Martin Gering, and Laura Conforti in Cell Reports. Published online December 10 2015 doi:10.1016/j.celrep.2015.11.032