Summary: Mutations in LRRK2 leads to dysfunctional auxilin and impaired synaptic vesicle endocytosis in Parkinson’s patients, researchers report.
Source: Northwestern University.
Northwestern Medicine scientists identified a cellular mechanism that leads to neurodegeneration in patients with Parkinson’s disease, according to a study published in Proceedings of the National Academy of Sciences.
Dimitri Krainc, MD, PhD, chair and Aaron Montgomery Ward Professor of Neurology, was the senior author of the study, which demonstrated a link between defective synaptic vesicle endocytosis and accumulation of toxic oxidized dopamine.
When it accumulates in the brain, oxidized dopamine has been shown to mediate the death of dopamine-containing neurons, causing the common motor symptoms observed in Parkinson’s disease (PD) patients, according to previous research conducted by Krainc and published in Science. While people naturally lose dopamine neurons as they age, patients with PD lose a much larger number of these neurons and the remaining cells are no longer able to compensate for the loss of brain function, leading to disease.
“In our prior work, we found that oxidized dopamine is toxic to neurons,” said Krainc, who is also director of the Center for Neurogenetics. “In this paper, we further explain how such oxidized dopamine is formed in synaptic terminals of neurons from patients with Parkinson’s disease.”
The process begins when a protein called auxilin is dysregulated by another protein, a mutated form of LRRK2. Normally, auxilin regulates the process of synaptic vesicle endocytosis, a mechanism that neurons use to replenish the chemical signals needed to communicate with each other.
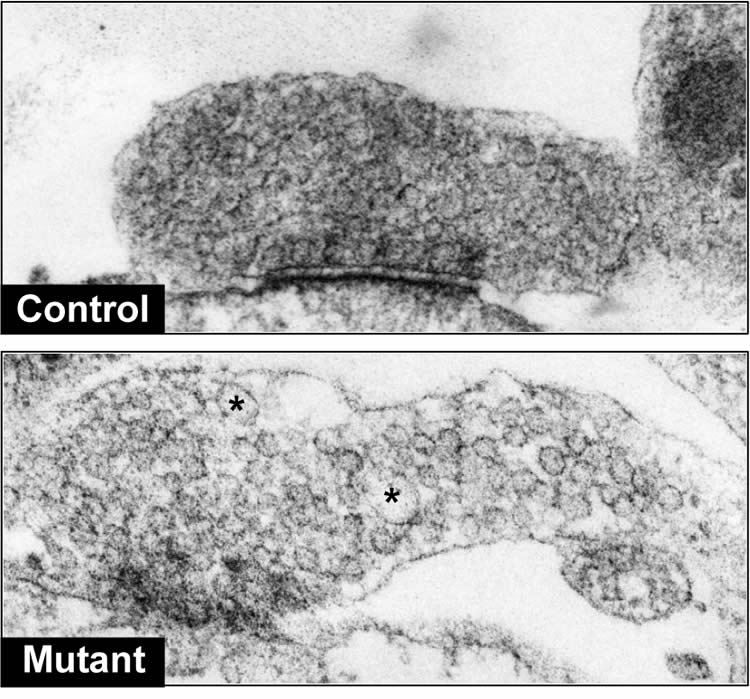
In the current study, scientists found that mutations in LRRK2 lead to dysfunctional auxilin and consequently impaired synaptic vesicle endocytosis. This manifests in inefficient packaging of dopamine into synaptic vesicles and an eventual buildup of dopamine in Parkinson’s neurons.
This pool of “extra” dopamine can be rapidly oxidized and become toxic to dopamine neurons, according to the study.
“These findings suggest that early therapeutic intervention in dysfunctional presynaptic terminals may prevent downstream toxic effects of oxidized dopamine and neurodegeneration in PD,” Krainc said.
In addition, these studies of genetic forms of Parkinson’s disease help identify converging pathways in the pathogenesis of sporadic and familial PD, highlighting the importance of investigating such cellular mechanisms to identify specific targets for therapy.
“This study is another example of how the emergence of genetic causes of Parkinson’s has helped us understand how disease develops and where to focus to identify key pathways and targets for drug development,” Krainc said.
Maria Nguyen, a fifth-year student in the Northwestern University Interdepartmental Neuroscience Program, was the first author of the study.
Funding:The study was supported by National Institutes of Health Grants R01 NS076054, R01 NS096240 and 2T32AG020506-16.
Source: Will Doss – Northwestern University
Publisher: Organized by NeuroscienceNews.com.
Image Source: NeuroscienceNews.com image is credited to Krainc et al.
Original Research: Abstract for “LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease” by Maria Nguyen and Dimitri Krainc in PNAS. Published May 7 2018.
doi:10.1073/pnas.1717590115
[cbtabs][cbtab title=”MLA”]Northwestern University “Early Synaptic Dysfunction Found in Parkinson’s Disease.” NeuroscienceNews. NeuroscienceNews, 24 May 2018.
<https://neurosciencenews.com/parkinsons-synaptic-dysfunction-9131/>.[/cbtab][cbtab title=”APA”]Northwestern University (2018, May 24). Early Synaptic Dysfunction Found in Parkinson’s Disease. NeuroscienceNews. Retrieved May 24, 2018 from https://neurosciencenews.com/parkinsons-synaptic-dysfunction-9131/[/cbtab][cbtab title=”Chicago”]Northwestern University “Early Synaptic Dysfunction Found in Parkinson’s Disease.” https://neurosciencenews.com/parkinsons-synaptic-dysfunction-9131/ (accessed May 24, 2018).[/cbtab][/cbtabs]
Abstract
LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease
Recently identified Parkinson’s disease (PD) genes involved in synaptic vesicle endocytosis, such as DNAJC6 (auxilin), have further implicated synaptic dysfunction in PD pathogenesis. However, how synaptic dysfunction contributes to the vulnerability of human dopaminergic neurons has not been previously explored. Here, we demonstrate that commonly mutated, PD-linked leucine-rich repeat kinase 2 (LRRK2) mediates the phosphorylation of auxilin in its clathrin-binding domain at Ser627. Kinase activity-dependent LRRK2 phosphorylation of auxilin led to differential clathrin binding, resulting in disrupted synaptic vesicle endocytosis and decreased synaptic vesicle density in LRRK2 patient-derived dopaminergic neurons. Moreover, impaired synaptic vesicle endocytosis contributed to the accumulation of oxidized dopamine that in turn mediated pathogenic effects such as decreased glucocerebrosidase activity and increased α-synuclein in mutant LRRK2 neurons. Importantly, these pathogenic phenotypes were partially attenuated by restoring auxilin function in mutant LRRK2 dopaminergic neurons. Together, this work suggests that mutant LRRK2 disrupts synaptic vesicle endocytosis, leading to altered dopamine metabolism and dopamine-mediated toxic effects in patient-derived dopaminergic neurons.