

Genetic analysis by Stowers investigators has implications for a genetic disorder known as Hirschsprung Syndrome.
Mutations in single genes can cause catastrophic diseases, such as Huntington’s Disease or sickle cell anemia. However, many conditions, including cancer, diabetes and birth defects are multigenic, arising from the collective failure of the function of more than one gene.
Researchers know that mutations in at least twelve individual genes are associated with the congenital defect Hirschprung Disease (HSCR), in which children are born lacking nerves that innervate the large intestine. Now two companion studies published in Human Molecular Genetics by Paul Trainor, Ph.D., Investigator at the Stowers Institute for Medical Research, identify a new gene associated with HSCR and show how the migration of cells that form the gut nervous system is impeded when the combined doses of two candidate genes are low. Understanding the genetic basis of HSCR offers hope for better diagnostics and treatment for it and other developmental defects caused by failure of cell migration.
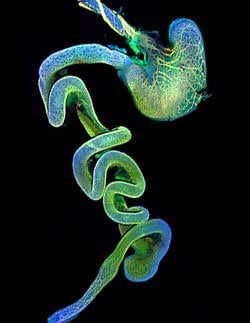
The cells that go awry in HSCR are a subset of what are called neural crest cells, embryonic cells that spring from the developing brain and spinal cord in mice or humans and then travel long distances to form, among others, structures in the face and heart, smooth muscle, and neurons of the peripheral nervous system, including those that innervate the gut.
Trainor has been interested in neural crest cells since he was a graduate student, often focusing on developmental defects caused by their malfunction. “Neural crest cells have to be born in the right place, migrate an incredibly long distance, survive the migration, multiply and then differentiate into a mature cell type,” says Trainor, who until recently was primarily interested in neural crest-related craniofacial anomalies. “In Hirschprung Disease they don’t make it to the end of the large intestine.”
As a result, the affected portion of the intestine cannot undergo peristaltic movements and waste products accumulate in the gut. Although surgery to remove the affected part of the gut improves the condition, some children are left with lasting complications including constipation, dehydration and poor nutrient absorption.
Both Human Molecular Genetics papers employ imaging techniques to visualize and track neural crest migration in mice mutant in HSCR candidate genes. The earlier study, published in January of 2012, reported that mice missing one of their two copies of a gene called Tcof1 not only produced fewer neural crest cells but that colonization of the gut by the remaining cells proceeded slowly compared to normal mice. Despite that delay, lagging cells eventually reached the end of the intestine before mice were born, just in time to innervate the gut properly.
“That paper showed that neural crest cells don’t seem to have a defined time window to colonize the entire gut as long as they do it by birth,” says Trainor, suggesting that for humans this is a good thing in terms of potential intervention. “If an embryo looked like it was going to have Hirschprung’s characteristics, that did not necessarily mean it would end up with that condition.”
If the first study showed that loss of a little Tcof1 had unusual but non-debilitating effects on gut innervation, the second paper, published in January of 2013, revealed what happens when you genetically chip away at the process. In the latter, the researchers eliminated one copy of Tcof1 along with one copy of another gene known to govern neural crest formation and proliferation, Pax3. Double mutant mice showed serious defects: fewer neural crest cells migrated out of the embryonic spinal cord, and the ones that remained underwent increased cell death as they traveled toward the gut. As a result, near the end of embryogenesis, Pax3/Tcof1 double mutant mice lacked the normal complement of neurons in the large intestine, a condition mimicking human HSCR.
“This is a much more dramatic result than the effect you might see following decreases in either gene alone,” says Amanda Barlow, Ph.D., a former Senior Research Associate in the Trainor lab and first author of both studies. “It’s like having two hammers which deliver two blows.”
Some molecular biologists might alter that metaphor slightly, to perhaps a hammer and crow bar, as Tcof1 and Pax3 are very different proteins. Pax3 is a “logical suspect”, a transcription factor that binds DNA and turns target genes known to stimulate cell proliferation off and on. By contrast, Tcof1 resides in a specialized cellular compartment called the nucleolus where the machinery that synthesizes all cellular proteins is constructed. How loss of a little of these strange bedfellows synergizes to promote developmental defects remains unknown. Trainor says it’s likely a threshold effect: losing a little Tcof1 might debilitate cellular growth just enough to make neural crest cells vulnerable to death if Pax3 doses were also compromised.
Barlow, now an Associate Scientist at University of Wisconsin, agrees, noting that researchers are increasingly aware that genetic interactions among seemingly unrelated genes are likely the norm, not the exception. “No gene exists in isolation,” she says. “To make an embryo the activity of many different pathways have to be integrated. There’s going to be a lot of cross-talk.”
Notes about this genetics research article
In addition to Trainor and Barlow, Mike Dixon, Ph.D., and Jill Dixon, Ph.D., of the University of Manchester in the UK contributed to both papers. Long-standing collaborators with Trainor, the Dixons were part of the team that first identified TCOF1 as responsible for Treacher Collins syndrome craniofacial anomalies, and they made the Tcof1 mouse model used in the these investigations.
The study was funded by the Stowers Institute for Medical Research, the March of Dimes, National Institute of Dental and Craniofacial Research (RO1 DE 016082-01) the National Institutes of Health (P50 DE 016215), the Healing Foundation and the Medical Research Council of the UK.
Contact: Paul Trainor, Ph.D – Stowers Institute for Medical Research
Source: Stowers Institute for Medical Research press release
Image Source: The image is credited to Naomi Tjaden, adapted from the Stowers Institute for Medical Research press release.
Original Research: Abstract for “Tcof1 acts as a modifier of Pax3 during enteric nervous system development and in the pathogenesis of colonic aganglionosis” by Amanda J. Barlow, Jill Dixon, Michael Dixon and Paul A. Trainor in Human Molecular Genetics. Published online January 2 2013 doi: 10.1093/hmg/dds528
Abstract for “Balancing neural crest cell intrinsic processes with those of the microenvironment in Tcof1 haploinsufficient mice enables complete enteric nervous system formation” by Amanda J. Barlow, Jill Dixon, Michael Dixon and Paul A. Trainor in Human Molecular Genetics. Published online January 6 2013 doi: 10.1093/hmg/ddr611




