Summary: Mice expressing a specific variant of the Scn1a gene associated with Dravet syndrome exhibited spontaneous seizures, disordered breathing and died prematurely.
Source: University of Connecticut
In sudden death in epilepsy, people stop breathing for no apparent reason and die. Now, a group of UConn neuroscientists have a lead as to why they report in the journal eLife.
“People with epilepsy have a high mortality rate, but it’s mysterious,” says Dan Mulkey, a neuroscientist in UConn’s physiology and neurobiology department.
More than one of every 1,000 people with epilepsy die each year from what’s called sudden unexpected death in epilepsy (SUDEP). No one knows why.
The explanation usually given is that the patient had a seizure that killed them. But seizures happen in the cortex, the top of the brain, and life-sustaining processes like breathing are controlled somewhere else entirely: the brainstem, the very bottom part of the brain that connects to the spinal cord. The two parts of the brain are quite distant from each other.
“It’s like, if the seizure is in New York, the brainstem is in San Francisco,” Mulkey says.
Many neurologists argue that a particularly bad seizure can travel down through the brain from the cortex to the brainstem to cause breathing or heartbeat malfunction, and that’s what kills in SUDEP. But Mulkey doesn’t buy it. People die of SUDEP without having an obvious seizure, and epilepsy patients can have breathing problems in the absence of seizures.
Instead, Mulkey and his colleagues, graduate students Fu-Shan Kuo and Colin Clearly, wondered if there was a genetic basis for SUDEP. Perhaps the same genetic mutation that causes the seizures also disrupts the cells in the brainstem that control breathing.
Kuo raised mice with the human mutation for a severe form of epilepsy called Dravet syndrome. Dravet syndrome is caused by mutations in a gene that shapes the channels through which sodium moves in and out of cells in the brain. If the sodium channels don’t function properly, cells can get overexcited. One cell’s overexcitement can travel through the brain like hysteria through a crowded stadium, stampeding into a seizure.
The gene mutated in Dravet syndrome is called sodium channel gene 1a, or Scn1a. It’s considered a super-culprit for epilepsy, with more than 1,200 different Scn1a mutations identified. The severity of the epilepsy caused by Scn1a depends on whether the mutation causes partial or complete loss of the sodium channel’s function. The Dravet mutation is on the severe end of the spectrum. People with Dravet syndrome tend to have dramatic seizures, exacerbated by hot weather, and the syndrome is very hard to control with anti-epileptic medications. SUDEP is sadly a frequent way for people with Dravet syndrome to die.
There’s a somewhat paradoxical part of Dravet syndrome, too: this Scn1a mutation makes the sodium channels less active, not more. Instead of making cells overactive, it makes them underactive. But there’s a catch. This mutation mostly affects inhibitory cells – that is, cells in charge of calming the brain down. They’re the stadium bouncers, so to speak. And if the bouncers are asleep on the job, the overexcited neurons can stampede uninhibited.
To understand how this might lead to SUDEP, Kuo wanted to test two things: first, whether the mice with the Dravet syndrome mutation show breathing problems and die prematurely of SUDEP, and second, whether the cells in the part of the mice’s brainstem that controls breathing were normal or were somehow perturbed by the mutation.
The first question was answered quickly: the mice with Dravet syndrome had bad seizures that became more severe when the mice got hot, exactly like humans with Dravet syndrome. They tended to die very young, in a manner similar to SUDEP; none lived much past three weeks.
The second question took longer to answer, but there were early clues that Kuo and Mulkey were on to something. The mice with Dravet Syndrome had disordered breathing. They tended to hypoventilate (breathe too little) for no apparent reason sometimes. Other times they would have long apneas, or pauses between breaths. And these mice didn’t breathe more in response to high carbon dioxide levels in the air, the way humans and normal mice do.
“We felt really good that our model was reflecting the human condition,” Mulkey says.
The next step was to actually look at the mice’s brainstems and see if something was wrong.
When Kuo zoomed in on the part of the brainstem that controls breathing, she saw that the inhibitory cells – the stadium bouncers of the brainstem – were definitely less active than they should have been. This led the excitatory neurons to run wild, and constantly tell the part of the brain that generates the breathing rhythm to push faster. But shouldn’t this lead to increased breathing, not stopping?
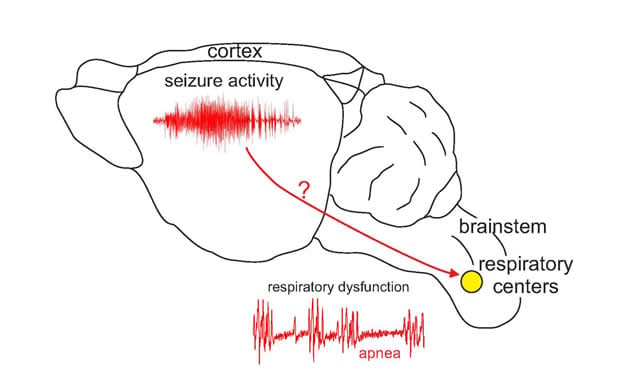
There is definitely something wrong with the breathing circuit in the brainstem in these mice, but Mulkey and Kuo cannot pinpoint the exact problem. So they’re still on the case. The next steps will be to look at mice that only express the Scn1a mutation in the brainstem or only in the cortex, and see if they also have problems. If mice with a mutation in the cortex but not the brainstem don’t have SUDEP, that would argue against the ‘seizure descending from cortex to brainstem’ hypothesis. The researchers also plan on looking at other parts of the breathing circuit to see whether other parts have gone haywire, too. Eventually, they hope to identify a key player that can be calmed – or prodded – to prevent the breathing system from breaking down, and ultimately save the lives of people with epilepsy.
Source:
University of Connecticut
Media Contacts:
Kim Krieger – University of Connecticut
Image Source:
The image is credited to Dan Mulkey and Virge Kask, University of Connecticut..
Original Research: Open access
“Disordered breathing in a mouse model of Dravet syndrome”. Fu-Shan Kuo, Colin M Cleary, Joseph J LoTurco, Xinnian Chen, Daniel K Mulkey Is a corresponding author.
eLife. doi:10.7554/eLife.43387
Abstract
Disordered breathing in a mouse model of Dravet syndrome
Dravet syndrome (DS) is a form of epilepsy with a high incidence of sudden unexpected death in epilepsy (SUDEP). Respiratory failure is a leading cause of SUDEP, and DS patients’ frequently exhibit disordered breathing. Despite this, mechanisms underlying respiratory dysfunction in DS are unknown. We found that mice expressing a DS-associated Scn1a missense mutation (A1783V) conditionally in inhibitory neurons (Slc32a1cre/+::Scn1aA1783V fl/+; defined as Scn1aΔE26) exhibit spontaneous seizures, die prematurely and present a respiratory phenotype including hypoventilation, apnea, and a diminished ventilatory response to CO2. At the cellular level in the retrotrapezoid nucleus (RTN), we found inhibitory neurons expressing the Scn1a A1783V variant are less excitable, whereas glutamatergic chemosensitive RTN neurons, which are a key source of the CO2/H+-dependent drive to breathe, are hyper-excitable in slices from Scn1aΔE26 mice. These results show loss of Scn1a function can disrupt respiratory control at the cellular and whole animal levels.