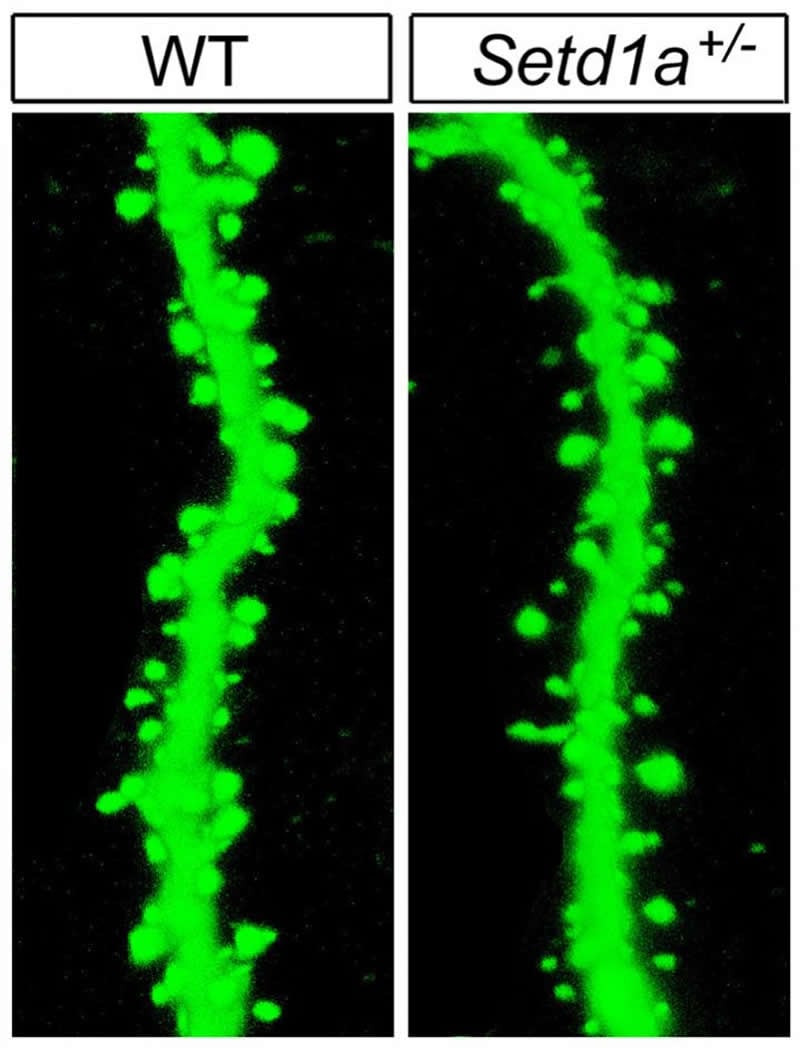
Summary: SETD1A, a gene associated with schizophrenia, stunts the growth and branching of dendrites and reduces the number of dendritic spines. Reinstating the normal expression of SETD1A in mouse models restored working memory function.
Source: NIH/NIMH
Researchers have discovered in mice how one of the few genes definitively linked to schizophrenia, called SETD1A, likely confers risk for the illness. Mice genetically engineered to lack a functioning version of the enzyme-coding gene showed abnormalities in working memory, mimicking those commonly seen in schizophrenia patients. Restoring the gene’s function corrected the working memory deficit. Counteracting the gene’s deficiencies also repaired neuronal circuit deficits in adult mice – suggesting clues for potential treatment strategies. A team of scientists led by Joseph Gogos, M.D., Ph.D., of Columbia University, New York City, reported on their research – supported by the National Institutes of Health – in Neuron.
“You could call SETD1A a master regulator,” explained David Panchision Ph.D., of the NIH’s National Institute of Mental Health (NIMH), which co-funded the study. “This schizophrenia risk gene codes for an enzyme that influences the expression of many other genes. In mice, a hobbled version of SETD1A disrupted gene expression in a network harboring other genomic suspects in schizophrenia. Remarkably, the resulting abnormalities were reversible.”
Researchers have identified both common and rare genetic variations that contribute to risk for schizophrenia. Mutant SETD1A is one of just a few rare genes known to unequivocally confer risk for schizophrenia. While common genetic variations linked to schizophrenia individually exert only tiny effects on risk, having just one mutant copy of SETD1A is sufficient to confer a large increase in disease risk. SETD1A plays a key role in epigenomic regulation – the switching on-and-off of genes in response to experience – a molecular process widespread in the brain. Mutations in SETD1A have primarily been found in people with schizophrenia, suggesting that this rare gene variation might hold important clues to the underlying disease process.
To find out how such a mutation in SETD1A affects brain cells, circuits, and behavior, Gogos and colleagues modeled the effects in mice carrying a mutation that halves the gene’s expression.
The genetically-altered mice faltered on tasks requiring navigation of a maze to receive a reward. The tasks test the animals’ working memory – holding information in mind and retrieving it to guide behavior – an ability often impaired in schizophrenia.
The mutated gene also disrupted the cellular machinery by which neurons communicate with each other. For example, it stunted the growth and branching of cell extensions and reduced the number of spines on these extensions, which are needed to relay chemical signals from neighboring cells into electrical impulses.
Underlying such impaired neuronal growth and function, the researchers discovered that the mutant SETD1A gene disrupted regulation of many other genes with which it is networked. Whole classes of genes were under-expressed while others were over-expressed, depending on their relationship to the gene. One class overlapped conspicuously with genetic variation associated with schizophrenia in key (pyramidal) neurons of the brain’s outer mantel, or cortex, with likely cumulative effects on cellular structure and function, suggest the researchers.
Experimentally reinstating normal expression of SETD1A in adult mice restored the animals’ working memory function. Moreover, inhibiting expression of a gene called LSD1, which counteracts SETD1A, corrected all of the animals’ behavioral and neuronal communication abnormalities. Evidence indicated that many of these mechanisms identified in mouse brain have been conserved through evolution and likely play similar roles in humans.
Reactivating SETD1A function or counteracting downstream effects of SETD1A deficiency in the adult brain, perhaps with LSD1 inhibitors, may hold promise for treating schizophrenia’s cognitive deficits, suggest the researchers.
“Although SETD1A mutations exist in a small percentage of all schizophrenia patients, many people diagnosed with the disorder have issues similar to those caused by this mutation,” explained Gogos. “Thus, therapies that are specific to SETD1A may indeed have wider implications for schizophrenia as a whole.”
Source:
NIH/NIMH
Media Contacts:
Jules Asher – NIH/NIMH
Image Source:
The image is credited to Jun Mukai/Gogos Lab/Columbia’s Zuckerman Institute.
Original Research: Closed access
“Recapitulation and Reversal of Schizophrenia-Related Phenotypes in Setd1a-Deficient Mice”. Mukai J, Cannavò E, Crabtree GW, Sun Z, Diamantopoulou A, Thakur P, Chang CY, Cai Y, Lomvardas S, Takata A, Xu B, Gogos JA.
Neuron doi:10.1016/j.neuron.2019.09.014.
Abstract
Recapitulation and Reversal of Schizophrenia-Related Phenotypes in Setd1a-Deficient Mice
Highlights
• Cognitive and circuitry deficits in a mouse model of SETD1A, a schizophrenia risk gene
• Reinstating Setd1a or antagonizing LSD1 activity in adulthood rescues deficits
• Setd1a binds promoters and enhancers with a striking overlap with Mef2 on enhancers
• Evolutionarily conserved Setd1a-bound enhancers may regulate psychiatric risk genes
Summary
SETD1A, a lysine-methyltransferase, is a key schizophrenia susceptibility gene. Mice carrying a heterozygous loss-of-function mutation of the orthologous gene exhibit alterations in axonal branching and cortical synaptic dynamics accompanied by working memory deficits. We show that Setd1a binds both promoters and enhancers with a striking overlap between Setd1a and Mef2 on enhancers. Setd1a targets are highly expressed in pyramidal neurons and display a complex pattern of transcriptional up- and downregulations shaped by presumed opposing functions of Setd1a on promoters and Mef2-bound enhancers. Notably, evolutionarily conserved Setd1a targets are associated with neuropsychiatric genetic risk burden. Reinstating Setd1a expression in adulthood rescues cognitive deficits. Finally, we identify LSD1 as a major counteracting demethylase for Setd1a and show that its pharmacological antagonism results in a full rescue of the behavioral and morphological deficits in Setd1a-deficient mice. Our findings advance understanding of how SETD1A mutations predispose to schizophrenia (SCZ) and point to novel therapeutic interventions.