Summary: A new study adds to the growing body of evidence that the origin of Huntington’s disease is rooted in childhood. Researchers say the HTT gene mutation affects both brain and body growth during development, and the increased susceptibility of brain cell death begins early in life.
Source: IOS Press
There is growing evidence to support the hypothesis that there is a neurodevelopmental component to the late-onset neurodegeneration occurring in the brain of huntingtin gene (HTT gene) mutation carriers, and that this increased susceptibility to brain cell death begins during childhood.
Experts discuss the evidence that the HTT gene mutation affects brain and body growth based on a unique study of children at risk for HD, the Kids-HD study, in a review paper and accompanying research article published in the Journal of Huntington’s Disease.
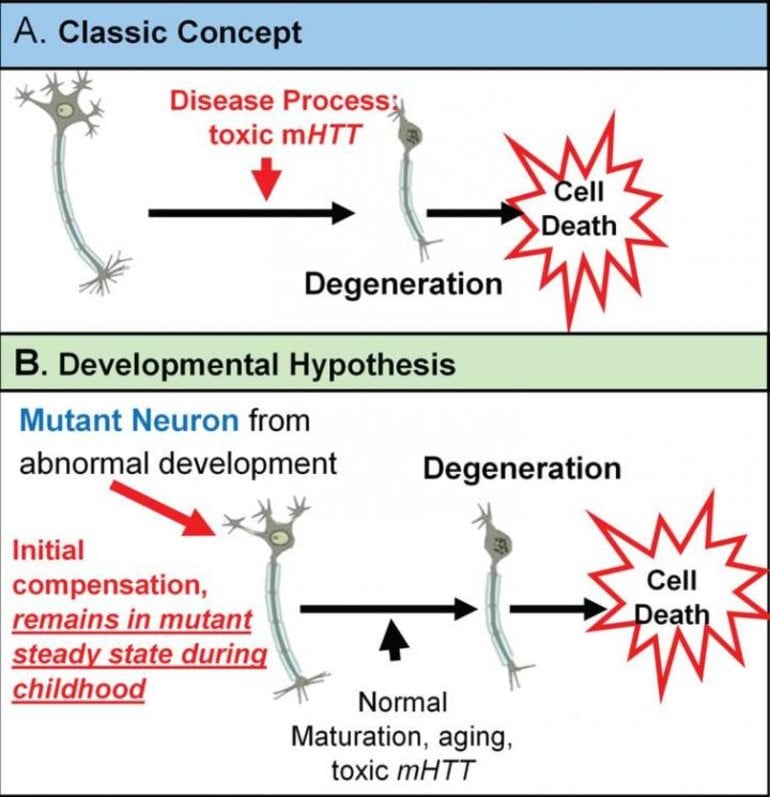
The classic concept is that Huntington’s disease is caused by toxic mutant huntingtin (mHTT) acting over time on mature brain cells. However, there is growing evidence for an alternative theory in which mHTT has an effect on brain development and that this altered development plays a vital role in the later degenerative process. This theory is based on the notion that wild-type huntingtin (HTT) function plays a role in normal brain development.
“Although the gene was discovered in 1993, we still don’t have a good understanding of what causes HD – how does the mutant gene cause brain cells to become ‘sick’ then die?” noted lead investigator Peg C. Nopoulos, MD, Department of Psychiatry, Department of Pediatrics, and Department of Neurology, University of Iowa Carver College of Medicine, Iowa City, IA, USA.
“The neurodevelopmental hypothesis is a relatively new way of thinking about the disease and can help focus research efforts in a new direction. This review and the accompanying study are important in reshaping our ideas about how we view the nature and timing of preventive treatment for HD and the factors that contribute to disease.”
The neurodevelopmental hypothesis of HD posits that the disease-causing gene mutation affects development of a specific region or specific brain circuit. These cells are abnormal in their growth; however, they are compensated for early in life. Therefore, despite abnormal development, there are no overt symptoms.
The abnormally developed cells remain in a “mutant steady state.” These cells are then vulnerable to dysfunction and degeneration later in life when they are subjected to stresses and strains, either normal (programmed synaptic elimination during puberty or through the aging process) or pathological (toxic effects of mHTT). In the end, the disease pathology results in neural degeneration with its accompanying cognitive and motor deficits.
The two papers focus on the most recent findings from Kids-HD, a unique brain imaging study of children aged six to 18 years old at risk for HD because they have a parent or grandparent with HD. The review discusses the effects of mHTT on brain development and the study evaluates the effect of mHTT on body development.
According to the authors, there is evidence in children as young as 6 who carry a mutation in the HTT gene, that production of mHTT alters the growth and development of the striatum and related circuitry. The gene contains a sequence of three DNA bases – cytosine-adenine-guanine (CAG) – repeated multiple times. The developmental changes appear to be influenced by the CAG repeat length and occur well before onset of symptoms of the disease. Deficits may then be compensated for by increased activity in other brain circuits, particularly those involving the cerebellum, and are manifest only when compensatory systems are no longer working.
The body development analysis used data from the 186 children in the Kids-HD study. Investigators applied simple measures of growth – height, weight, and the combined BMI measure – to compare changes in two groups – those who carried the CAG repeat expansion mutation in the HTT gene and those who did not.
Around puberty the study began to show an altered trajectory of growth in HTT gene mutation carriers. The pace at which their BMI increased slowed over time so that at by about 17 years old, that group had substantially lower BMI than the group without the gene mutation. Boys with the gene mutation tended to be taller than the control group, but with lower weight; girls with the gene mutation tended to be around the same height, but lower in weight.
Importantly, although the gene mutation carriers were roughly 30 years from the expected time of onset of the disease, the mutant HTT gene had already affected their growth and development. This work is important because it suggests that the mutation is altering the body even before the onset of neurological disease in midlife.
Gene therapy trials are currently underway to evaluate the effectiveness of drugs to slow disease progression in affected individuals, and future trials will ultimately aim to prevent disease onset by delivery of gene therapy to gene mutation carriers – those with mHTT, but no symptoms.
“Gene therapy trials are finally here. However, interfering with a gene responsible for brain development early in life must be done with an abundance of caution,” commented Dr. Nopoulos. “Understanding how mHTT affects brain development is vital in the context of planning disease prevention therapies.”
HD is a fatal genetic neurodegenerative disease that causes the progressive breakdown of nerve cells in the brain. An estimated 250,000 people in the United States are either diagnosed with, or at risk for, the disease. Symptoms include personality changes, mood swings and depression, forgetfulness and impaired judgment, unsteady gait, and involuntary movements (chorea). Every child of an HD parent has a 50% chance of inheriting the gene. Patients usually survive 10 – 20 years after diagnosis.
About this Huntington’s disease research news
Source: IOS Press
Contact: Press Office – IOS Press
Image: The image is credited to Journal of Huntington’s Disease.
Original Research: Open access.
“Developmental Trajectory of Height, Weight, and BMI in Children and Adolescents at Risk for Huntington’s Disease: Effect of mHTT on Growth” by Nopoulos et al. Journal of Huntington’s Disease.
Abstract
Developmental Trajectory of Height, Weight, and BMI in Children and Adolescents at Risk for Huntington’s Disease: Effect of mHTT on Growth
Background:
The gene (Huntingtin or HTT) causing Huntington’s disease (HD) is vital for development and is expressed throughout the brain and body lifelong. The mutant form (mHTT) may influence growth and development.
Objective:
To determine the impact of mHTT on human measures of growth, including height, weight, and body mass index (BMI), between child and adolescent carriers of mHTT and control peers.
Methods:
Children ages 6–18 years of age (n = 186) at risk for HD were enrolled in the KidsHD study. For research purposes only, genetic testing was performed to classify participants as Gene-Expanded (GE = 78) or as Gene Non-Expanded (GNE = 108). Outcome measures included height, weight, and body mass index (BMI). Mixed models were used to determine if non-linear age trends differed between groups for BMI, height, and weight.
Results:
Differences were seen in the trajectory of BMI in which the GE group reached a plateau in late adolescence with no further increase, compared with a nearly linear increase in the GNE group. There was a significant sex interaction pattern where GE males were taller than GNE males in adolescence, in the presence of similar weight. In contrast, GE females weighed significantly less than their GNE counterparts in adolescence, in the presence of similar height.
Conclusion:
Measures of growth are abnormal in child and adolescent carriers of mHTT, decades before HD onset. Although further studies are needed for replication, the current findings suggest that developmental aberrations may be systemic and a vital part of disease pathology.