

Summary: Findings indicate a large part of the genetic risk of Alzheimer’s involves a microglial response to amyloid-beta.
Source: VIB Flanders
Our DNA determines a large part of our risk for Alzheimer’s disease, but it remained unclear how many genetic risk factors contribute to disease. A team led by Prof. Bart De Strooper (VIB-KU Leuven) and Dr. Mark Fiers now show that many risk factors affect brain maintenance cells called microglia, and more particularly their response to amyloid-beta, one of the proteins aggregating in the brains of Alzheimer patients. The individual effects of small genetic variations are likely small, but the combination of hundreds of such subtle alterations might tip the balance and cause disease.
Why do some people get Alzheimer’s disease while others do not, even when growing very old? Despite decades of research, we still don’t know the full answer to this question. Epidemiological studies show that about two-thirds of a person’s risk for Alzheimer’s disease is genetically determined. A few dozen risk genes have been identified, however, recent evidence shows that there could be hundreds of additional genetic variants that each contribute in a small but significant way to disease risk.
From risk gene to disease mechanism
Bart De Strooper (VIB-KU Leuven) has been studying the mechanisms of Alzheimer’s disease for decades. His team tries to find out what this combined genetic risk can teach us about how the disease develops in our brain: “Two crucial questions arise from the myriad of genetic studies. First, what is the link between these Alzheimer’s risk genes and the amyloid-beta plaques or tau tangles we find in Alzheimer’s brains; and second, are they all involved in one central cellular or molecular pathway, or do they define many parallel pathways that all lead to Alzheimer’s?”
The researchers set out to understand when these genes are expressed and in particular, whether they respond to tau or amyloid-beta pathology. “When it comes to risk, you always need to take the context into account,” explained Mark Fiers, co-lead author of the study. “If you don’t wear your seatbelt in the car, there is no problem as long as you don’t have an accident.”
With this in mind, the researchers aimed to understand under which circumstances genetic risk for Alzheimer’s comes into play. Fiers: “Almost every person develops some degree of Alzheimer pathology in the brain, i.e. amyloid-beta plaques and tau tangles. However, some people remain cognitively healthy despite a high pathology load, while others develop Alzheimer’s symptoms quite rapidly.”
“To gain more insight we checked gene expression in two different mouse models of Alzheimer’s, one displaying amyloid-beta and the other tau pathology, at different ages,” says Annerieke Sierksma, a postdoctoral researcher in De Strooper’s lab. “We identified that many of the genes linked to Alzheimer’s risk are particularly responsive to amyloid-beta but not to tau pathology.”
Microglia activation
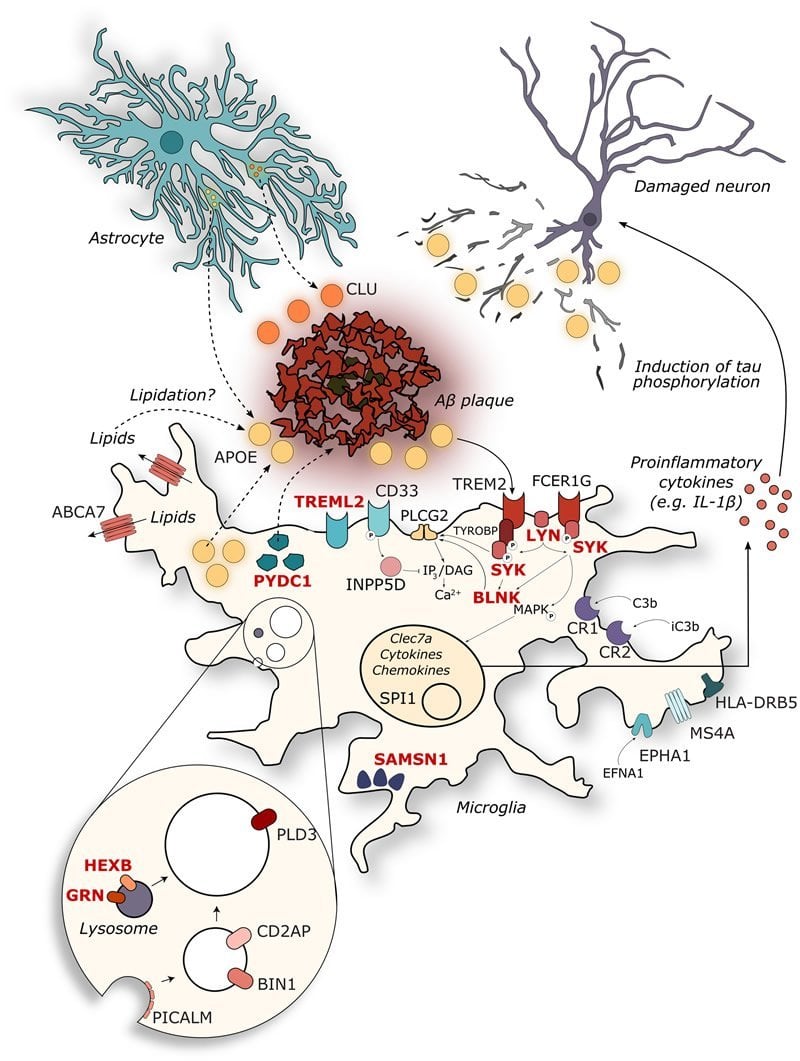
The team identified 11 new risk genes that are significantly upregulated when facing increased amyloid-beta levels. All these genes are expressed in microglia, cells that play a key role in brain maintenance.
Ashley Lu, a PhD student closely involved in the analysis: “We could confirm that microglia exposed to amyloid-beta drastically switch to an activated status, something that occurs to a much lesser extent in the tau mice. These new insights indicate that a large part of the genetic risk of Alzheimer’s disease involves the microglial response to amyloid-beta.”
Understanding genetic risk
Should we rethink the classical gene-based view, where certain mutations or genetic variants lead to disease? De Strooper thinks so: “One single genetic variant within a functional network will not lead to disease. However, multiple variants within the same network may tip the balance to a disease-causing disturbance. Such a hypothesis could also explain the conundrum that some /individuals with a lot of amyloid-beta in their brain do not develop clinical symptoms.”
“While amyloid-beta might be the trigger of the disease, it is the genetic make-up of the microglia, and possibly other cell types, which determines whether a pathological response is induced,” adds Fiers. “Identifying which genetic variants are crucial to such network disturbances and how they lead to altered gene expression will be the next big challenge.”
Why mice?
“Profiling of postmortem brain tissue only provides insights into the advanced stages of the disease and does not allow to delineate cause-consequence relationships,” explains De Strooper. “Genetically modified mouse models on the other hand only partially recapitulate the disease, but they allow for detailed insights into the initial steps of disease, which is of high relevance for preventative therapeutic interventions.”
Questions from patients
A breakthrough in research is not the same as a breakthrough in medicine. The realizations of VIB researchers can form the basis of new therapies, but the development path still takes years. This can raise a lot of questions.
Source:
VIB Flanders
Media Contacts:
Katrina Wright – VIB Flanders
Image Source:
The image is credited to Annerieke Sierksma et al.
Original Research: Open access
“Novel Alzheimer risk genes determine the microglia response to amyloid‐β but not to TAU pathology”. Sierksma, Lu et al.
Sierksma, Lu et al doi:10.15252/emmm.201910606.
Abstract
Novel Alzheimer risk genes determine the microglia response to amyloid‐β but not to TAU pathology
Polygenic risk scores have identified that genetic variants without genome‐wide significance still add to the genetic risk of developing Alzheimer’s disease (AD). Whether and how subthreshold risk loci translate into relevant disease pathways is unknown. We investigate here the involvement of AD risk variants in the transcriptional responses of two mouse models: APPswe/PS1L166P and Thy‐TAU22. A unique gene expression module, highly enriched for AD risk genes, is specifically responsive to Aβ but not TAU pathology. We identify in this module 7 established AD risk genes (APOE, CLU, INPP5D, CD33, PLCG2, SPI1, and FCER1G) and 11 AD GWAS genes below the genome‐wide significance threshold (GPC2, TREML2, SYK, GRN, SLC2A5, SAMSN1, PYDC1, HEXB, RRBP1, LYN, and BLNK), that become significantly upregulated when exposed to Aβ. Single microglia sequencing confirms that Aβ, not TAU, pathology induces marked transcriptional changes in microglia, including increased proportions of activated microglia. We conclude that genetic risk of AD functionally translates into different microglia pathway responses to Aβ pathology, placing AD genetic risk downstream of the amyloid pathway but upstream of TAU pathology.




