

UA researchers have identified a molecular defect in motor neurons that may help explain the mechanisms underlying ALS, or Lou Gehrig’s Disease.
By now, most everyone has seen videos all over social media of friends and family dousing themselves in ice cold water as part of the ALS Ice Bucket Challenge.
The 2014 campaign was part of an effort to raise awareness of amyotrophic lateral sclerosis, or ALS, a devastating neurodegenerative disorder that causes progressive muscle weakening and loss of coordination. But what made much less of a splash in the media is what researchers are doing to tackle the issue.
ALS is notoriously difficult to treat, and relatively little is known about exactly how and why it occurs. In a rare discovery, a clear molecular defect has been found at the junctions between neurons and muscles, which may provide greater insight into the fundamental mechanisms of ALS, according to a new study by Daniela Zarnescu, associate professor in the University of Arizona’s Department of Molecular and Cellular Biology, and Alyssa Coyne, a graduate student in the UA’s Neuroscience Graduate Interdisciplinary Program and first author on the study. The paper is published in the Journal of Neuroscience.
In healthy people, nerve cells called motor neurons make contact with muscle fibers at places called neuromuscular junctions, which allows for appropriate control of movement and other critical functions. In ALS patients, motor neurons die off in droves, preventing these connections from occurring.
To study ALS, Zarnescu and Coyne use the fruit fly Drosophila melanogaster as their model, which gives the researchers the advantage of molecular and genetic approaches that allow them to more easily pinpoint exactly when and where things go wrong.
“When you tell people that you use the fruit fly as a model of human disease, you get some funny looks,” Zarnescu said. “But using simplistic models can help you uncover what’s really important in the context of the disease.”
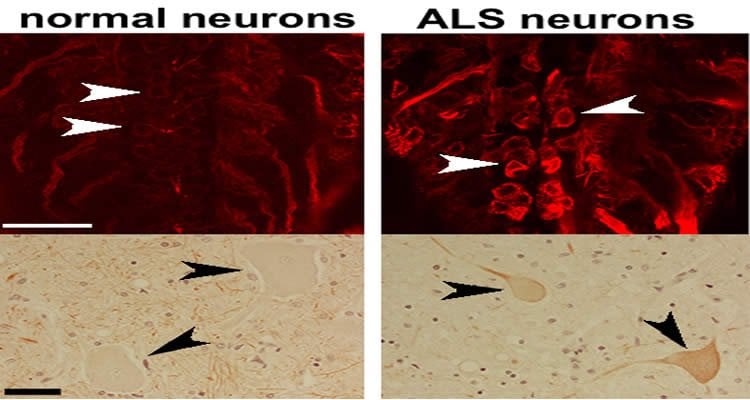
Zarnescu and Coyne studied a protein called TDP-43, which previously has been implicated in ALS. The team found that TDP-43 regulates the creation and transport of another protein called Futsch at the neuromuscular junction. In the ALS model, TDP-43 prevents Futsch from making it to the neuromuscular junction, which results in a faulty connection.
“Alyssa discovered that this particular molecule is not regulated properly. It’s not made in the right place or in the right amount,” Zarnescu said. “Instead of being transported to the neuromuscular junction, it stays in the body of the cell and can’t maintain the stability of the connection.”
The researchers then wanted to determine if increasing the amount of Futsch protein would help repair the poor connection. Astoundingly, overexpressing Futsch in motor neurons had the effect of increasing the stability of the connection, increasing the lifespan of motor neurons and restoring motor function in the ALS fruit flies.
At this point, you might be asking: What does ALS in the nervous system of the fly have to do with ALS in humans?
To find out, Zarnescu and Coyne collaborated with researchers at the Barrow Neurological Institute in Phoenix to look at cells from the spinal cords of human ALS patients. The team looked at a protein called MAP1B, which is the mammalian version of the Futsch protein. Remarkably, the localization of MAP1B was altered in a very similar manner to the Futsch protein in fruit flies. The similarities suggest comparable defects in both human and fly models of ALS.
“This highlights the importance of studying human disease in simple models,” Zarnescu said. “These models are extremely powerful, and predictive of defects that occur in human patients.”
According to Zarnescu and Coyne, the findings represent a major step forward in understanding and eventually treating the disease.
“This study is among the first to highlight such a clear molecular defect at synaptic connections in ALS,” Zarnescu said. “We don’t yet know exactly what is going on in ALS, but this discovery provides a possible explanation.”
Contact: Raymond Sanchez – University of Arizona
Source: University of Arizona press release
Image Source: The image is credited to Daniela Zarnescu and is adapted from the University of Arizona press release
Original Research: Abstract for “Futsch/MAP1B mRNA Is a Translational Target of TDP-43 and Is Neuroprotective in a Drosophila Model of Amyotrophic Lateral Sclerosis” by Alyssa N. Coyne, Bhavani Bagevalu Siddegowda, Patricia S. Estes, Jeffrey Johannesmeyer, Tina Kovalik, Scott G. Daniel, Antony Pearson, Robert Bowser, and Daniela C. Zarnescu in Journal of Neuroscience. Published online November 26 2014 doi:10.1523/JNEUROSCI.2526-14.2014




