NIH-funded mouse study identifies therapeutic target for clearing out toxic proteins damaged during neurodegenerative disorders.
A study of mice shows how proteasomes, a cell’s waste disposal system, may break down during Alzheimer’s disease (AD), creating a cycle in which increased levels of damaged proteins become toxic, clog proteasomes, and kill neurons. The study, published in Nature Medicine and supported by the National Institutes of Health, suggests that enhancing proteasome activity with drugs during the early stages of AD may prevent dementia and reduce damage to the brain.
“This exciting research advances our understanding of the role of the proteasomes in neurodegeneration and provides a potential way to alleviate symptoms of neurodegenerative disorders,” said Roderick Corriveau, Ph.D., program director at the NIH’s National Institute of Neurological Disorders and Stroke (NINDS), which provided funding for the study.
The proteasome is a hollow, cylindrical structure which chews up defective proteins into smaller, pieces that can be recycled into new proteins needed by a cell. To understand how neurodegenerative disorders affect proteasomes, Natura Myeku, Ph.D., a postdoctoral fellow working with Karen E. Duff, Ph.D., professor of pathology and cell biology at Columbia University, New York City, focused on tau, a structural protein that accumulates into clumps called tangles in the brain cells of patients with AD and several other neurodegenerative disorders known as tauopathies.
Using a genetically engineered mouse model of tauopathy, as well as looking at cells in a dish, the scientists discovered that as levels of abnormal tau increased, the proteasome activity slowed down.
Treating the mice at the early stages of tauopathy with the drug rolipram increased proteasome activity, decreased tau accumulations and prevented memory problems. They found that the drug worked exclusively during the early stages degeneration, which began around four months of age. It helped four-month old tauopathy mice remember the location of hidden swimming platforms as well as control mice, and better than tauopathy mice that received placebos. Treating mice at later stages of the disease was not effective.
“These results show, for the first time, that you can activate the proteasome in the brain using a drug and effectively slow down the disease, or prevent it from taking a hold,” said Dr. Duff, senior author of the study.
Rolipram was initially developed as an antidepressant but is not used clinically due to its side effects. It increases the levels of cyclic AMP, a compound that triggers many reactions inside brain cells. Rolipram works by blocking cyclic AMP phosphodiesterase four (PDE4), an enzyme that degrades cyclic AMP. The scientists found that cyclic AMP levels are critical for controlling proteasome activity. Treating brain slices from tauopathy mice with rolipram, or a version of cyclic AMP that PDE4 cannot degrade, reduced the accumulation of tau and sped proteasome activity.
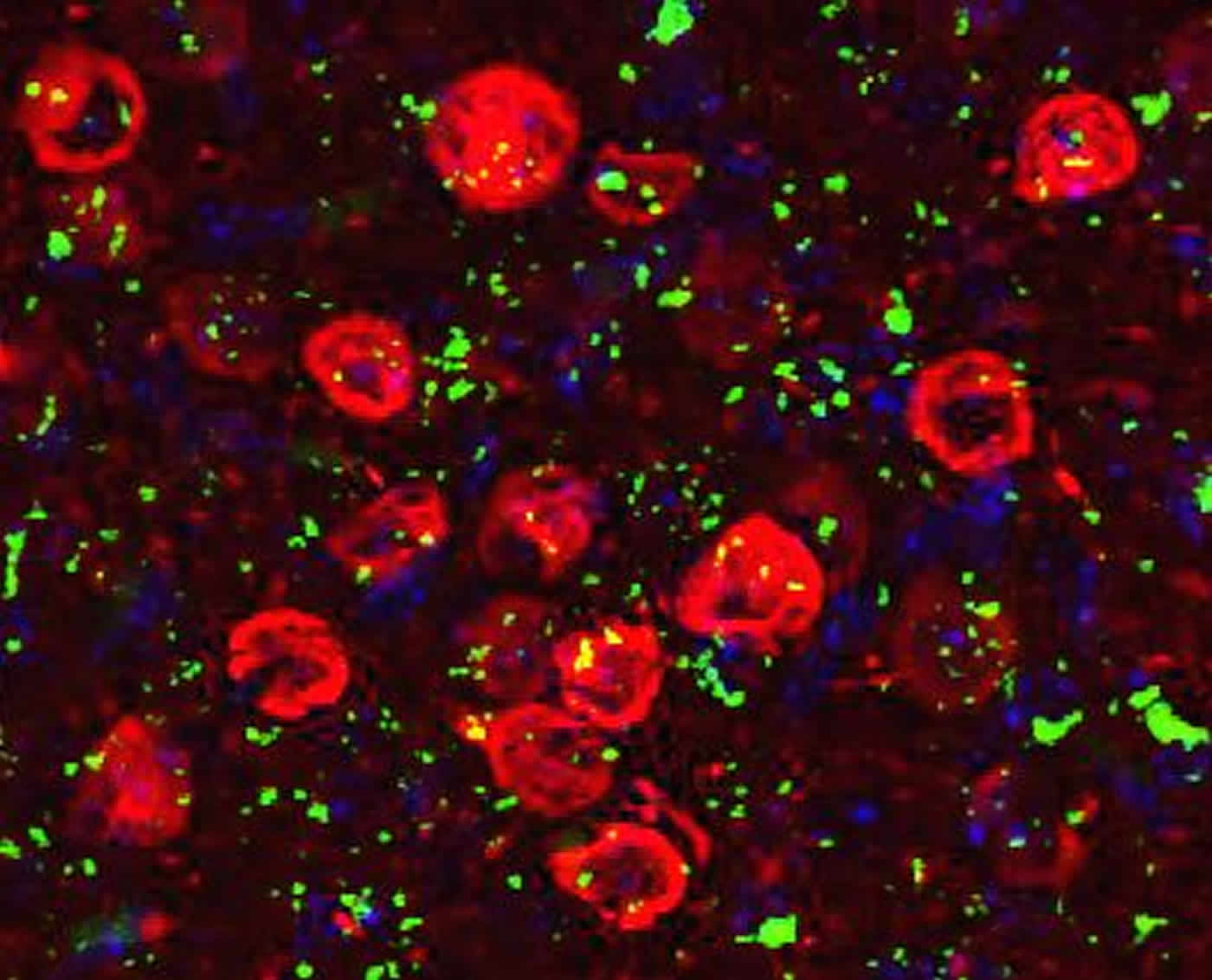
“We were hoping to show, using rolipram, that increasing cyclic AMP is a pharmaceutical strategy worth pursuing. The suggestion is not that rolipram should immediately go into the clinic but that drugs with mechanisms similar to rolipram should be investigated further,” said Dr. Myeku.
Drs. Myeku and Duff plan to further investigate proteasome activity and the impact of tau and other disease-related proteins on this system for chewing up and clearing out damaged proteins. In addition, they want to search libraries of FDA-approved compounds or new molecules for drugs that work in a similar way to rolipram or activate proteasomes by different pathways.
“The proteasome system we are studying also degrades proteins associated with a number of other neurodegenerative diseases such as Parkinson’s, Huntington’s, frontotemporal degeneration and amyotrophic lateral sclerosis. We may be able to apply these findings to other disorders that accumulate proteins,” said Dr. Duff.
Funding: This work was supported by the NINDS (NS074593) and the CurePSP Foundation.
Source: Barbara McMakin and Christopher G. Thomas – NIH/NINDS
Image Source: The image is credited to Karen Duff, Ph.D., Columbia University.
Original Research: Abstract for “Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling” by Natura Myeku, Catherine L Clelland, Sheina Emrani, Nikolay V Kukushkin, Wai Haung Yu, Alfred L Goldberg and Karen E Duff in Nature Medicine. Published online December 21 2015 doi:10.1038/nm.4011
Abstract
Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling
The ubiquitin proteasome system (UPS) degrades misfolded proteins including those implicated in neurodegenerative diseases. We investigated the effects of tau accumulation on proteasome function in a mouse model of tauopathy and in a cross to a UPS reporter mouse (line Ub-G76V-GFP). Accumulation of insoluble tau was associated with a decrease in the peptidase activity of brain 26S proteasomes, higher levels of ubiquitinated proteins and undegraded Ub-G76V-GFP. 26S proteasomes from mice with tauopathy were physically associated with tau and were less active in hydrolyzing ubiquitinated proteins, small peptides and ATP. 26S proteasomes from normal mice incubated with recombinant oligomers or fibrils also showed lower hydrolyzing capacity in the same assays, implicating tau as a proteotoxin. Administration of an agent that activates cAMP–protein kinase A (PKA) signaling led to attenuation of proteasome dysfunction, probably through proteasome subunit phosphorylation. In vivo, this led to lower levels of aggregated tau and improvements in cognitive performance.
“Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling” by Natura Myeku, Catherine L Clelland, Sheina Emrani, Nikolay V Kukushkin, Wai Haung Yu, Alfred L Goldberg and Karen E Duff in Nature Medicine. Published online December 21 2015 doi:10.1038/nm.4011