

Summary: Activity of the c-Fos gene begins the increase on the ninth day of exposure to antidepressants. By day 14, mice showed behavioral changes associated with SSRI use. This may explain why suicide rates drop after nine days of treatment, and symptoms of depression improve after three weeks.
Source: Rockefeller University
Some highly effective medications also happen to be highly mysterious. Such is the case with the antidepressant drugs known as selective serotonin reuptake inhibitors, or SSRIs: They are the most common treatment for major depression and have been around for more than 40 years, yet scientists still do not know exactly how they work.
Nor is it known why only two out of every three patients respond to SSRI treatment, or why it typically takes several weeks for the drugs to take effect—a significant shortcoming when you’re dealing with a disabling mood disorder that can lead to impaired sleep, loss of appetite, and even suicide.
New research by a team of Rockefeller scientists helps elucidate how SSRIs combat depression. Their work, published in Molecular Psychiatry, could one day make it possible to predict who will respond to SSRIs and who will not, and to reduce the amount of time it takes for the drugs to act.
Brain Teasers
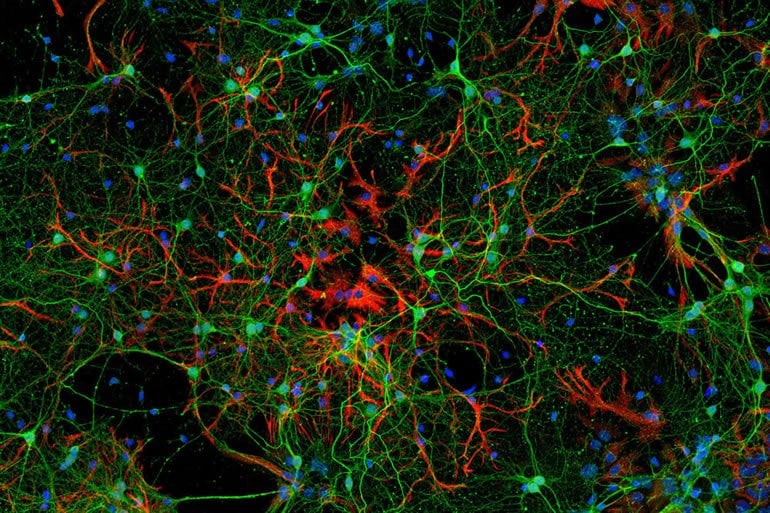
Major depression—also known as clinical depression—is firmly rooted in biology and biochemistry. The brains of people who suffer from the disease show low levels of certain neurotransmitters, the chemical messengers that allow neurons to communicate with one another. And studies have linked depression to changes in brain volume and impaired neural circuitry.
Scientists have long known that SSRIs rapidly increase the available amount of the neurotransmitter serotonin, leading to changes that go well beyond brain chemistry: Research suggests the drugs help reverse the neurological damage associated with depression by boosting the brain’s innate ability to repair and remodel itself, a characteristic known as plasticity. Yet the molecular details of how SSRIs work their magic remains a mystery.
Researchers in the late Paul Greengard’s Laboratory of Molecular and Cellular Neuroscience set out to trace the chain of molecular events triggered by one of the most widely prescribed and effective SSRIs: fluoxetine, also known as Prozac. In particular, they wanted to see if they could tie the effects of the drug to specific changes in gene expression.
Led by senior research associate Revathy Chottekalapanda, the team treated mice with fluoxetine for 28 days, then measured the animals’ biochemical and behavioral responses to the drug. They conducted these experiments in a mouse strain prone to anxiety that offered several advantages including the fact that “improvement in anxiety, in addition to depressive-like behavior, is a good measure of antidepressant drug response,” Chottekalapanda says.
Using a combination of behavioral tests and real-time RNA analysis, the researchers were able to monitor changes in the animals’ behavior and gene expression as they responded to the drug. Things started to get interesting on the ninth day of treatment: The activity of a gene called c-Fos began to increase markedly, and by day 14 the mice were showing telltale behavioral changes—they were moving around more, for example, and took an interest in food even after being moved to a new environment. The timing was remarkable, Chottekalapanda says, because it aligned with well-established milestones in the treatment of human patients.
“For example, the rate of suicide drops after nine days of treatment, and people tend to feel better after two to three weeks,” she says.
As it turns out, c-Fos helps create a so-called transcription factor, AP-1, which activates specific genes by binding to their DNA. The sudden appearance of these molecules therefore raised several new questions: Which genes does AP-1 activate? What triggers the production of AP-1 in the first place? And how does this whole sequence of events ultimately beat back depression?
Domino Effect
Chottekalapanda and her colleagues began by analyzing the mice’s cortex, a brain region previously shown to be essential for the antidepressant response, looking for changes in genes and DNA-binding proteins to which AP-1 might possibly bind. Focusing especially on the nine-day mark, they found changes in mouse genes whose human counterparts have been linked to depression and antidepressant drug response.
Walking the cat backward, the team was able to identify specific molecules known as growth factors that spur the manufacture AP-1 itself, and the pathways through which they act. Taken together, these results painted a detailed picture of how fluoxetine and other SSRIs work.
First, the drugs ramp up the amount of serotonin available in the brain. This triggers a molecular chain reaction that ultimately makes brain cells increase their AP-1 production—an effect that only begins to take off on day nine of treatment. AP-1 then switches on several genes that promote neuronal plasticity and remodeling, allowing the brain to reverse the neurological damage associated with depression. After two to three weeks, the regenerative effects of those changes can be seen—and felt.
“For the first time, we were able to put a number of molecular actors together at the crime scene in a time- and sequence-specific manner,” says Chottekalapanda.
To confirm their molecular model of SSRI response, Chottekalapanda and her team gave mice treated with fluoxetine an additional substance designed to block one of the pathways necessary to the production of AP-1.
The results were striking: When the researchers prevented the mice from producing AP-1, the effects of the drug were severely blunted. Moreover, gene-expression analysis showed that blocking the formation of AP-1 partially reversed the activation of some of the genes responsible for the antidepressant response.
One Word: Plasticity
The implications of the team’s findings could be far-reaching. For example, the genes that AP-1 targets could be used as biomarkers to predict whether a given patient will respond to SSRI treatment. And the cast of molecular characters involved in fluoxetine response could potentially be targeted with drugs to improve the efficacy of antidepressant treatment, perhaps even reducing the amount of time it takes for SSRIs to kick in.
Towards that end, Chottekalapanda is already conducting experiments to clarify the precise reasons for the nine-day delay in AP-1 production. She would also like to know whether these molecular players are mutated or inactive in people who don’t respond to SSRIs, and to discover precisely how the genes that AP-1 regulates in response to fluoxetine would promote neuronal plasticity.
That last piece of the puzzle might prove to be especially important. Depression is not the only disorder that could potentially be remedied by enlisting the brain’s innate healing abilities: Chottekalapanda suspects that the same plasticity-promoting pathways that are activated by antidepressants such as fluoxetine could potentially be used to treat other neurological and neurodegenerative conditions such as Alzheimer’s and Parkinson’s disease.
“If we can figure out what they do, we can potentially develop treatments to reverse the damage involved not only with depression but also with other neurological disorders,” she says.
About this psychopharmacology research article
Source:
Rockefeller University
Contacts:
Press Office – Rockefeller University
Image Source:
The image is credited to Rockefeller University.
Original Research: Open access
“AP-1 controls the p11-dependent antidepressant response” by Revathy U. Chottekalapanda et al. Molecular Psychiatry.
Abstract
AP-1 controls the p11-dependent antidepressant response
Selective serotonin reuptake inhibitors (SSRIs) are the most widely prescribed drugs for mood disorders. While the mechanism of SSRI action is still unknown, SSRIs are thought to exert therapeutic effects by elevating extracellular serotonin levels in the brain, and remodel the structural and functional alterations dysregulated during depression. To determine their precise mode of action, we tested whether such neuroadaptive processes are modulated by regulation of specific gene expression programs. Here we identify a transcriptional program regulated by activator protein-1 (AP-1) complex, formed by c-Fos and c-Jun that is selectively activated prior to the onset of the chronic SSRI response. The AP-1 transcriptional program modulates the expression of key neuronal remodeling genes, including S100a10 (p11), linking neuronal plasticity to the antidepressant response. We find that AP-1 function is required for the antidepressant effect in vivo. Furthermore, we demonstrate how neurochemical pathways of BDNF and FGF2, through the MAPK, PI3K, and JNK cascades, regulate AP-1 function to mediate the beneficial effects of the antidepressant response. Here we put forth a sequential molecular network to track the antidepressant response and provide a new avenue that could be used to accelerate or potentiate antidepressant responses by triggering neuroplasticity.




