Summary: Rockefeller University researchers reveal Huntington’s neurons are much larger than healthy cells.
Source: Rockefeller University.
With new findings, scientists may be poised to break a long impasse in research on Huntington’s disease, a fatal hereditary disorder for which there is currently no treatment.
One in 10,000 Americans suffer from the disease, and most begin to show symptoms in middle age as they develop jerky movements—and as these patients increasingly lose brain neurons, they slide into dementia. But the new research suggests that these symptoms may be a late manifestation of a disease that originates much earlier, in the first steps of embryonic development.
A team at Rockefeller led by Ali Brivanlou, the Robert and Harriet Heilbrunn Professor, developed a system to model Huntington’s in human embryonic stem cells for the first time. In a report published in Development, they describe early abnormalities in the way Huntington’s neurons look, and how these cells form larger structures that had not previously been associated with the disease.
“Our research supports the idea that the first domino is pushed soon after fertilization,” Brivanlou says, “and that has consequences down the line. The final domino falls decades after birth, when the symptoms are observable.”
The findings have implications for how to best approach treating the disorder, and could ultimately lead to effective therapies.
A new tool
Huntington’s is one of the few diseases with a straightforward genetic culprit: One hundred percent of people with a mutated form of the Huntingtin (HTT) gene develop the disease. The mutation takes the form of extra DNA, and causes the gene to produce a longer-than-normal protein. The DNA itself appears in the form of a repeating sequence, and the more repeats there are, the earlier the disease sets in.
Research on Huntington’s has thus far relied heavily on animal models of the disease, and has left many key questions unanswered. For example, scientists have not been able to resolve what function the HTT gene serves normally, or how its mutation creates problems in the brain.
Suspecting that the disease works differently in humans, whose brains are much bigger and more complex than those of lab animals, Brivanlou, along with research associates Albert Ruzo and Gist Croft, developed a cell-based human system for their research. They used the gene editing technology CRISPR to engineer a series of human embryonic stem cell lines, which were identical apart from the number of DNA repeats that occurred at the ends of their HTT genes.
“We started seeing things that were completely unexpected,” says Brivanlou. “In cell lines with mutated HTT, we saw giant cells. It looked like a jungle of disorganization.”
When cells divide, they typically each retain one nuclei. However, some of these enlarged, mutated cells flaunted up to 12 nuclei—suggesting that neurogenesis, or the generation of new neurons, was affected. The disruption was directly proportional to how many repeats were present in the mutation: The more repeats there were, the more multinucleated neurons appeared.
“Our work adds to the evidence that there is an unrecognized developmental aspect to the pathology,” Brivanlou says. “Huntington’s may not be just a neurodegenerative disease, but also a neurodevelopmental disease.”
Toxic or essential?
Treatments for Huntington’s have typically focused on blocking the activity of the mutant HTT protein, the assumption being that the altered form of the protein was more active than normal, and therefore toxic to neurons. However, Brivanlou’s work shows that the brain disruption may actually be due to a lack of HTT protein activity.
To test its function, the researchers created cell lines that completely lacked the HTT protein. These cells turned out to be very similar to those with Huntington’s pathology, corroborating the idea that a lack of the protein—not an excess of it—is driving the disease.
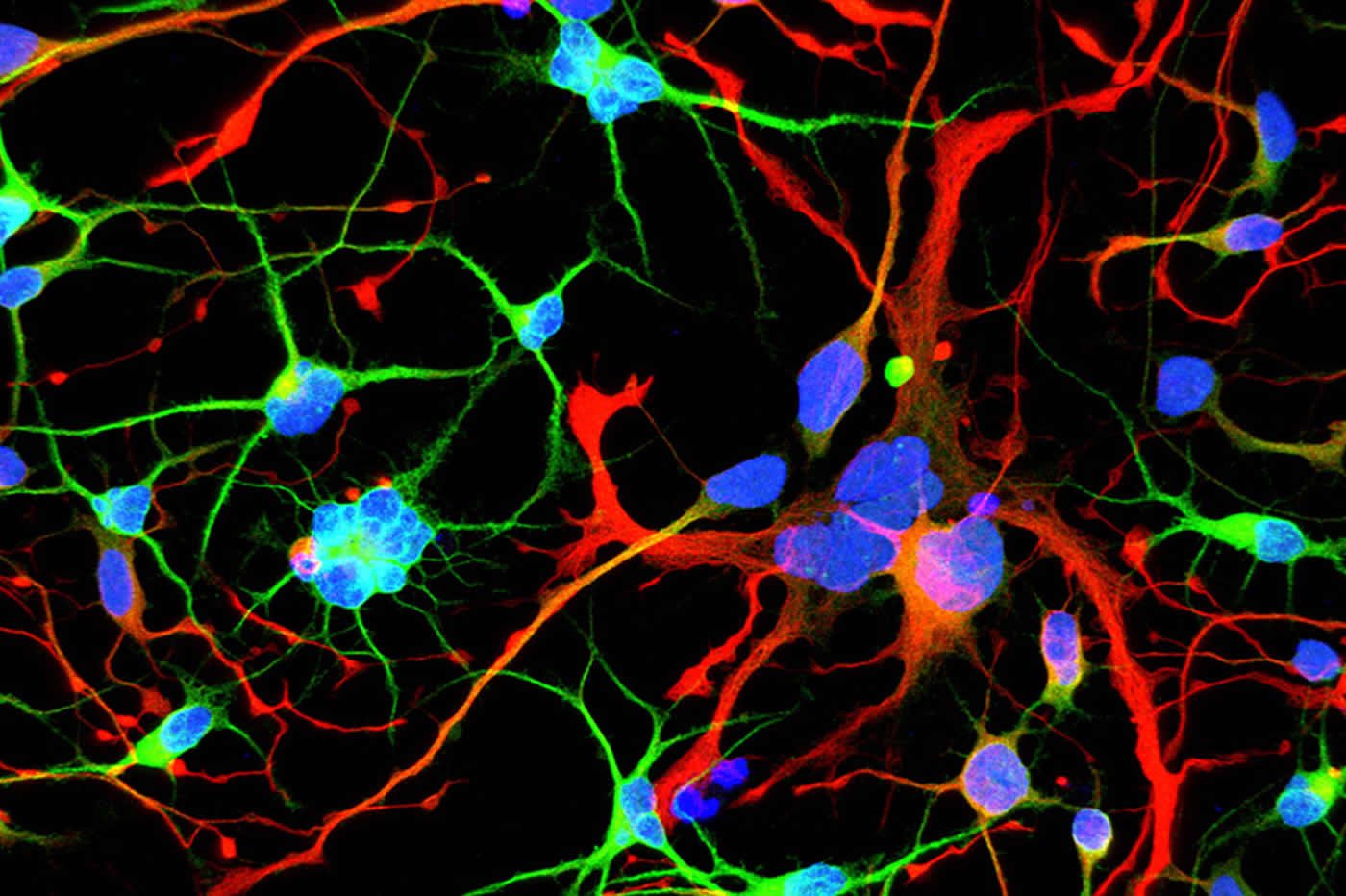
The findings are significant, Brivanlou notes, since they indicate that existing treatments that were designed to block HTT activity may actually do more harm than good.
“We should rethink our approach to treating Huntington’s,” he says. “Both the role of the HTT protein and the timing of treatment need to be reconsidered; by the time a patient is displaying symptoms, it may be too late to medicate. We need to go back to the earliest events that trigger the chain reaction that ultimately results in disease so we can focus new therapies on the cause, not the consequences.”
The researchers hope their new cell lines will be a useful resource for studying the cellular and molecular intricacies of Huntington’s further, and suggest they may provide a model for examining other diseases of the brain that are specific to humans.
Source: Katherine Fenz – Rockefeller University
Publisher: Organized by NeuroscienceNews.com.
Image Source: NeuroscienceNews.com image is adapted from the Rockefeller University news release.
Original Research: Abstract in Development.
doi:10.1242/dev.156844
[cbtabs][cbtab title=”MLA”]Rockefeller University “Uncovering the Early Origins of Huntington’s Disease.” NeuroscienceNews. NeuroscienceNews, 29 January 2018.
<https://neurosciencenews.com/huntingtons-origin-8389/>.[/cbtab][cbtab title=”APA”]Rockefeller University (2018, January 29). Uncovering the Early Origins of Huntington’s Disease. NeuroscienceNews. Retrieved January 29, 2018 from https://neurosciencenews.com/huntingtons-origin-8389/[/cbtab][cbtab title=”Chicago”]Rockefeller University “Uncovering the Early Origins of Huntington’s Disease.” https://neurosciencenews.com/huntingtons-origin-8389/ (accessed January 29, 2018).[/cbtab][/cbtabs]
Abstract
Chromosomal instability during neurogenesis in Huntington’s disease
Huntington’s disease (HD) is a fatal neurodegenerative disease caused by expansion of CAG repeats in the Huntingtin gene (HTT). Neither its pathogenic mechanisms nor the normal functions of HTT are well understood. To model HD in humans, we engineered a genetic allelic series of isogenic human embryonic stem cell (hESC) lines with graded increases in CAG repeat length. Neural differentiation of these lines unveiled a novel developmental HD phenotype: the appearance of giant multinucleated telencephalic neurons at an abundance directly proportional to CAG repeat length, generated by a chromosomal instability and failed cytokinesis over multiple rounds of DNA replication. We conclude that disrupted neurogenesis during development is an important, unrecognized aspect of HD pathogenesis. To address the function of normal HTT protein we generated HTT+/− and HTT−/− lines. Surprisingly, the same phenotype emerged in HTT−/− but not HTT+/− lines. We conclude that HD is a developmental disorder characterized by chromosomal instability that impairs neurogenesis, and that HD represents a genetic dominant-negative loss of function, contrary to the prevalent gain-of-toxic-function hypothesis. The consequences of developmental alterations should be considered as a new target for HD therapies.