

Summary: Researchers have decoded the 3D structure of the Huntingtin protein. The discovery may help develop new treatments for Huntington’s disease.
Source: Max Planck Institute.
Mutations on a single gene, the huntingtin gene, are the cause of Huntington’s disease. They lead to an incorrect form of the correspondent protein. With the help of cryo-electron microscopy researchers from the Max Planck Institute of Biochemistry in Martinsried and Ulm University have now decoded the three-dimensional, molecular structure of the healthy human huntingtin protein. This now enables its functional analysis. An improved understanding of the structure and the function of the huntingtin protein could contribute to the development of new treatment methods in the future.
Huntington’s disease often begins with emotional disturbances and is characterized by involuntary muscle movement and loss of mental ability. Until today, the neurological disease is one of the hereditary diseases still incurable and fatal. The protein HTT, also called huntingtin, plays the central role in Huntington’s disease. For 25 years it has been known that mutations of the huntingtin gene, which is the blueprint for the same protein, cause Huntington’s disease.
Although researchers have worked on it for many years, there are still many hurdles to overcome. Now Rubén Fernández-Busnadiego from the Max Planck Institute of Biochemistry and Stefan Kochanek, head of the Department of Gene Therapy at the University Hospital Ulm, have succeeded in decoding the molecular, three-dimensional structure of the huntingtin protein.
Overcome hurdle
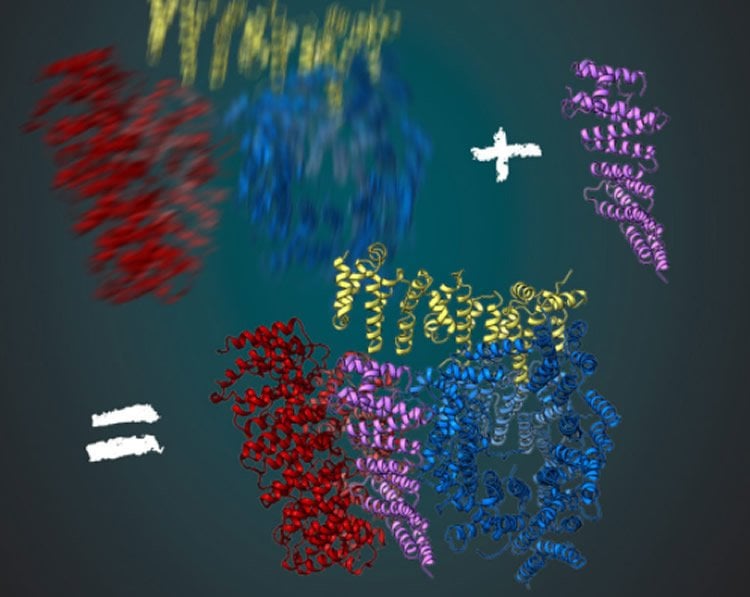
Stefan Kochanek and his team have been working on the production and purification of huntingtin for a long time. What has prevented a detailed analysis of the protein in recent decades? Fernández-Busnadiego, an expert in cryo-electron microscopy, mentions two main factors: “First of all, cryo-electron microscopy has only been optimized in recent years to look at protein structures with almost molecular resolution. The second reason is that the huntingtin protein is very flexible in its structure. Just now, we have found also a solution for this problem.” During the analysis, pictures of the protein are being taken from different perspectives under the microscope. The three-dimensional molecular structure can be computed from the large number of resulting images. For this, the protein must always be in the same conformation. Fernández-Busnadiego explains: “This would be similar to a person being photographed in the dark. If the person does not stand still for a while, the shot will be blurry.”
To get a clear picture, the researchers in the Kochanek lab have been looking for other proteins that interact with huntingtin and stabilize it. That worked for the protein HAP40. “Huntingtin in connection with HAP40 is stabilized in a particular conformation. Thus, averaged over many pictures, we were able to derive the three-dimensional structure,” said Kochanek. “If we remain in the analogy of the photo in the dark, then the protein acts like a chair for the photographed person. Sitting on it, the person moves much less and the image is much sharper with the same exposure time,” adds Fernández-Busnadiego.
Hope for new treatments
“Although we have known for some time that the mutation of the huntingtin gene has severe consequences, we still know relatively little about the function and tasks of the healthy protein,” explains Kochanek. Proteins are the molecular machines of the cell. In order to fulfil their versatile tasks, they have a certain three-dimensional structure, similar to a specific component in a machine. “Now that we know the exact structure of huntingtin, we can further study which areas of huntingtin are particularly important and how other proteins cooperate with huntingtin functionally. In this way structures could be deduced to be targeted therapeutically by certain drugs.”
Right now, there is a lot going on in Huntington’s disease research. Great hope is directed to a method, which mutes the huntingtin gene with so-called Antisense-Oligonucleotides for the treatment of Huntington’s disease. These small molecules reduce the formation of huntingtin proteins in the cells, but the drug cannot differentiate between the normal and the pathologically altered huntingtin protein. Also for this reason, it is important to learn more about the function of the healthy huntingtin protein. Kochanek confidently looks into the future: “The decoded structure will bring us a big step ahead”.
“The antisense oligonucleotide treatment trials are presently only conducted at very few clinics. The Neurological University Clinic Ulm leads the drug testing for Germany. Some of the currently reviewed antisense oligonucleotides reduce both the formation of the normal and the mutant huntingtin protein, while others predominantly seek to lower the altered huntingtin. It is currently unclear whether a partial inhibition of the formation of normal huntingtin protein has disadvantages and is tolerated without undesirable side effects. This is yet another reason why it is important to learn more about the normal function of the huntingtin protein. To this end, the study will make an important contribution,” says Bernhard Landwehrmeyer, the Director of the Huntington Outpatient Clinic of the Neurology Department at the University Medical Center Ulm and heads the world’s largest international cohort study on Huntington’s disease.
Source: Christiane Menzfeld – Max Planck Institute
Publisher: Organized by NeuroscienceNews.com.
Image Source: NeuroscienceNews.com image is credited to MPI of Biochemistry/ Illustration: Gabriele Stautner, ARTIFOX.
Original Research: Abstract in Nature.
doi:10.1038/nature25502
[cbtabs][cbtab title=”MLA”]Max Planck Institute “Decoding the Structure of Huntintin.” NeuroscienceNews. NeuroscienceNews, 23 February 2018.
<https://neurosciencenews.com/huntingtin-structure-8550/>.[/cbtab][cbtab title=”APA”]Max Planck Institute (2018, February 23). Decoding the Structure of Huntintin. NeuroscienceNews. Retrieved February 23, 2018 from https://neurosciencenews.com/huntingtin-structure-8550/[/cbtab][cbtab title=”Chicago”]Max Planck Institute “Decoding the Structure of Huntintin.” https://neurosciencenews.com/huntingtin-structure-8550/ (accessed February 23, 2018).[/cbtab][/cbtabs]
Abstract
The cryo-electron microscopy structure of huntingtin
Huntingtin (HTT) is a large (348 kDa) protein that is essential for embryonic development and is involved in diverse cellular activities such as vesicular transport, endocytosis, autophagy and the regulation of transcription1,2. Although an integrative understanding of the biological functions of HTT is lacking, the large number of identified HTT interactors suggests that it serves as a protein–protein interaction hub1,3,4. Furthermore, Huntington’s disease is caused by a mutation in the HTT gene, resulting in a pathogenic expansion of a polyglutamine repeat at the amino terminus of HTT5,6. However, only limited structural information regarding HTT is currently available. Here we use cryo-electron microscopy to determine the structure of full-length human HTT in a complex with HTT-associated protein 40 (HAP40; encoded by three F8A genes in humans)7 to an overall resolution of 4 Å. HTT is largely α-helical and consists of three major domains. The amino- and carboxy-terminal domains contain multiple HEAT (huntingtin, elongation factor 3, protein phosphatase 2A and lipid kinase TOR) repeats arranged in a solenoid fashion. These domains are connected by a smaller bridge domain containing different types of tandem repeats. HAP40 is also largely α-helical and has a tetratricopeptide repeat-like organization. HAP40 binds in a cleft and contacts the three HTT domains by hydrophobic and electrostatic interactions, thereby stabilizing the conformation of HTT. These data rationalize previous biochemical results and pave the way for improved understanding of the diverse cellular functions of HTT.




