

Doctors commonly recommend patients increase their intake of calcium as a means of combating osteoporosis for aging bones. However, calcium also plays an essential role in the neurological functioning of the brain, where it must be tightly regulated and not rise to excessive levels. A signaling molecule, calcium enables learning, cognition and the retention of memories. Calcium also facilitates communication among nerve cells and transports molecules to the many branches of the nerve cell.
Building on scientific evidence implicating disturbed calcium regulation in brain aging accumulated through the past 30 years, a research team in the University of Kentucky Department of Pharmacology and Nutritional Sciences led by principal investigator Philip Landfield has found a connection between unhealthy brain aging and a protein responsible for regulating calcium at the molecular level, called FKBP1b. The team’s groundbreaking research, which was published July 29 in the Journal of Neuroscience, identifies a molecular mechanism occurring within the cell that appears to cause unhealthy brain aging. The research suggests the absence or addition of the FKBP1b protein is a strong determinant of functioning in the hippocampus region, a part of the brain responsible for cognition and memory retention.
Unhealthy brain aging is defined as a reduction in brain function resulting in memory impairment. Excess calcium in brain cells appears responsible for important aspects of unhealthy brain aging, and may also increase susceptibility to diseases such as Alzheimer’s, ALS, Parkinson’s and vascular dementia. Until now, the precise molecular cause of the disturbed calcium regulation in brain aging has remained unknown to scientists.
After learning about the FKBP1b protein’s recently uncovered role in the heart, the UK researchers wondered whether FKBP1b in the hippocampus region declines with brain aging. They then found evidence of reduced FKBP1b gene expression with aging in the hippocampus. This discovery prompted the researchers at the University of Kentucky to test whether boosting FKBP1b in the hippocampus region could reverse or prevent brain aging linked to memory loss.
“It is well-recognized that normal aging is the greatest risk factor for Alzheimer’s disease, but nobody knows why,” Landfield, a professor in the department, said. “It’s possible this (decreased FKBP1b) is the missing link.”
The team used an advanced gene therapy approach to inject harmless virus particles, which created additional copies of the FKBP1b protein, into the hippocampus of aging rats. The memory abilities of three groups of rats were tested two months after the injections. One group of young rats received a control injection, one group of aged rats received a control injection and one aged group received an injection of the FKBP1b-producing virus particles. The aged group with raised levels of FKBP1b showed restored calcium regulation and dramatically improved cognitive function, allowing them to perform the memory task as well as or better than the young rats. In addition, the researchers have repeated and extended the results in a subsequent study being prepared for publication.
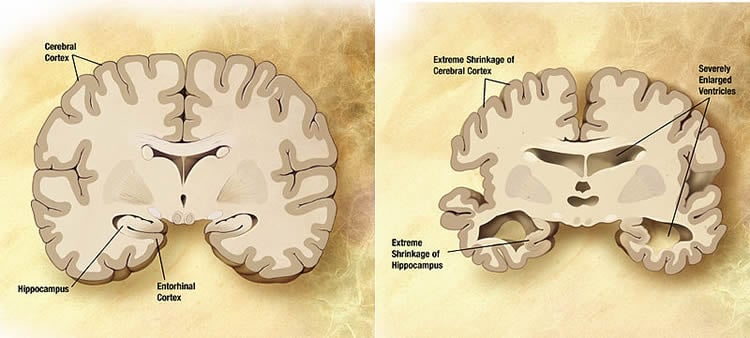
The research provides evidence the manifestations of brain aging can be reversed, and cognition and memory function restored, by altering levels of FKBP1b. This finding is also significant for Alzheimer’s patients as the researchers found a decline in the FKBP1b protein in the hippocampus of people who had early-stage Alzheimer’s. The research has implications for preventing brain aging associated with the progression of Alzheimer’s, and opens the door for pharmaceutical development aimed at sustaining levels of FKBP1b and keeping calcium in check.
“We showed FKBP1b is a master regulator of calcium in brain cells, and when we restore it, it restores the regulation of calcium and dramatically improves learning in the aged animals,” Landfield said. “In all my years of doing research, I’ve never seen a compound this effective; it’s rare that tests of a hypothesis satisfy each of the criteria that have to be met.”
The UK team is the only known group studying FKBP1b in brain aging. As a next step, the researchers are interested in investigating why FKBP1b declines with age. Landfield said there is promise to regulate the protein through Vitamin D, which is known to restore calcium deficiencies in other cells.
Funding: The research was supported by a grant from the National Institute on Aging.
Source: Elizabeth Adams – University of Kentucky
Image Source: The image is credited to the NIH and is in the public domain
Original Research: Full open access research for “Reversal of Aging-Related Neuronal Ca2+ Dysregulation and Cognitive Impairment by Delivery of a Transgene Encoding FK506-Binding Protein 12.6/1b to the Hippocampus” by John C. Gant, Kuey-Chu Chen, Inga Kadish, Eric M. Blalock, Olivier Thibault, Nada M. Porter, and Philip W. Landfield in Journal of Neuroscience. Published online July 15 2015 doi:10.1523/JNEUROSCI.1248-15.2015
Abstract
Reversal of Aging-Related Neuronal Ca2+ Dysregulation and Cognitive Impairment by Delivery of a Transgene Encoding FK506-Binding Protein 12.6/1b to the Hippocampus
Brain Ca2+ regulatory processes are altered during aging, disrupting neuronal, and cognitive functions. In hippocampal pyramidal neurons, the Ca2+-dependent slow afterhyperpolarization (sAHP) exhibits an increase with aging, which correlates with memory impairment. The increased sAHP results from elevated L-type Ca2+ channel activity and ryanodine receptor (RyR)-mediated Ca2+ release, but underlying molecular mechanisms are poorly understood. Previously, we found that expression of the gene encoding FK506-binding protein 12.6/1b (FKBP1b), a small immunophilin that stabilizes RyR-mediated Ca2+ release in cardiomyocytes, declines in hippocampus of aged rats and Alzheimer’s disease subjects. Additionally, knockdown/disruption of hippocampal FKBP1b in young rats augments neuronal Ca2+ responses. Here, we test the hypothesis that declining FKBP1b underlies aging-related hippocampal Ca2+ dysregulation. Using microinjection of adeno-associated viral vector bearing a transgene encoding FKBP1b into the hippocampus of aged male rats, we assessed the critical prediction that overexpressing FKBP1b should reverse Ca2+-mediated manifestations of brain aging. Immunohistochemistry and qRT-PCR confirmed hippocampal FKBP1b overexpression 4–6 weeks after injection. Compared to aged vector controls, aged rats overexpressing FKBP1b showed dramatic enhancement of spatial memory, which correlated with marked reduction of sAHP magnitude. Furthermore, simultaneous electrophysiological recording and Ca2+ imaging in hippocampal neurons revealed that the sAHP reduction was associated with a decrease in parallel RyR-mediated Ca2+ transients. Thus, hippocampal FKBP1b overexpression reversed key aspects of Ca2+ dysregulation and cognitive impairment in aging rats, supporting the novel hypothesis that declining FKBP1b is a molecular mechanism underlying aging-related Ca2+ dysregulation and unhealthy brain aging and pointing to FKBP1b as a potential therapeutic target.
SIGNIFICANCE STATEMENT This paper reports critical tests of a novel hypothesis that proposes a molecular mechanism of unhealthy brain aging and possibly, Alzheimer’s disease. For more than 30 years, evidence has been accumulating that brain aging is associated with dysregulation of calcium in neurons. Recently, we found that FK506-binding protein 12.6/1b (FKBP1b), a small protein that regulates calcium, declines with aging in the hippocampus, a brain region important for memory. Here we used gene therapy approaches and found that raising FKBP1b reversed calcium dysregulation and memory impairment in aging rats, allowing them to perform a memory task as well as young rats. These studies identify a potential molecular mechanism of brain aging and may also have implications for treatment of Alzheimer’s disease.
“Reversal of Aging-Related Neuronal Ca2+ Dysregulation and Cognitive Impairment by Delivery of a Transgene Encoding FK506-Binding Protein 12.6/1b to the Hippocampus” by John C. Gant, Kuey-Chu Chen, Inga Kadish, Eric M. Blalock, Olivier Thibault, Nada M. Porter, and Philip W. Landfield in Journal of Neuroscience. Published online July 15 2015 doi:10.1523/JNEUROSCI.1248-15.2015




