Summary: The FDA have approved the first drug designed to treat patients with Duchenne muscular dystrophy.
Source: FDA.
The U.S. Food and Drug Administration today approved Exondys 51 (eteplirsen) injection, the first drug approved to treat patients with Duchenne muscular dystrophy (DMD). Exondys 51 is specifically indicated for patients who have a confirmed mutation of the dystrophin gene amenable to exon 51 skipping, which affects about 13 percent of the population with DMD.
“Patients with a particular type of Duchenne muscular dystrophy will now have access to an approved treatment for this rare and devastating disease,” said Janet Woodcock, M.D., director of the FDA’s Center for Drug Evaluation and Research. “In rare diseases, new drug development is especially challenging due to the small numbers of people affected by each disease and the lack of medical understanding of many disorders. Accelerated approval makes this drug available to patients based on initial data, but we eagerly await learning more about the efficacy of this drug through a confirmatory clinical trial that the company must conduct after approval.”
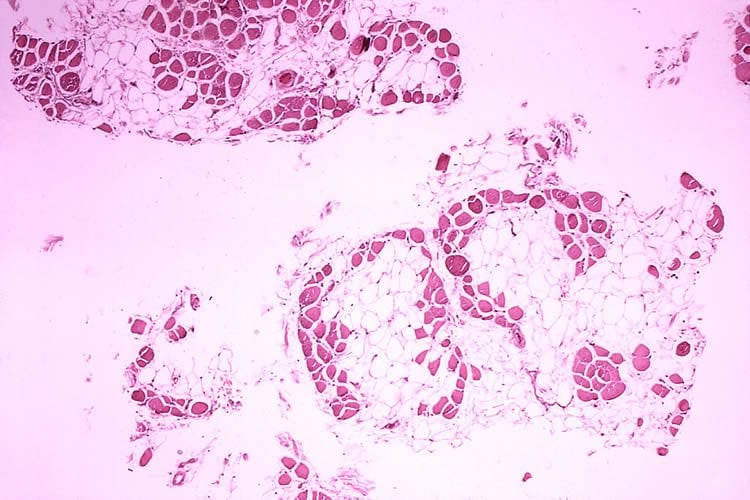
DMD is a rare genetic disorder characterized by progressive muscle deterioration and weakness. It is the most common type of muscular dystrophy. DMD is caused by an absence of dystrophin, a protein that helps keep muscle cells intact. The first symptoms are usually seen between three and five years of age, and worsen over time. The disease often occurs in people without a known family history of the condition and primarily affects boys, but in rare cases it can affect girls. DMD occurs in about one out of every 3,600 male infants worldwide.
People with DMD progressively lose the ability to perform activities independently and often require use of a wheelchair by their early teens. As the disease progresses, life-threatening heart and respiratory conditions can occur. Patients typically succumb to the disease in their 20s or 30s; however, disease severity and life expectancy vary.
Exondys 51 was approved under the accelerated approval pathway, which provides for the approval of drugs that treat serious or life-threatening diseases and generally provide a meaningful advantage over existing treatments. Approval under this pathway can be based on adequate and well-controlled studies showing the drug has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit to patients (how a patient feels or functions or whether they survive). This pathway provides earlier patient access to promising new drugs while the company conducts clinical trials to verify the predicted clinical benefit.
The accelerated approval of Exondys 51 is based on the surrogate endpoint of dystrophin increase in skeletal muscle observed in some Exondys 51-treated patients. The FDA has concluded that the data submitted by the applicant demonstrated an increase in dystrophin production that is reasonably likely to predict clinical benefit in some patients with DMD who have a confirmed mutation of the dystrophin gene amenable to exon 51 skipping. A clinical benefit of Exondys 51, including improved motor function, has not been established. In making this decision, the FDA considered the potential risks associated with the drug, the life-threatening and debilitating nature of the disease for these children and the lack of available therapy.
Under the accelerated approval provisions, the FDA is requiring Sarepta Therapeutics to conduct a clinical trial to confirm the drug’s clinical benefit. The required study is designed to assess whether Exondys 51 improves motor function of DMD patients with a confirmed mutation of the dystrophin gene amenable to exon 51 skipping. If the trial fails to verify clinical benefit, the FDA may initiate proceedings to withdraw approval of the drug.
The most common side effects reported by participants taking Exondys 51 in the clinical trials were balance disorder and vomiting.
The FDA granted Exondys 51 fast track designation, which is a designation to facilitate the development and expedite the review of drugs that are intended to treat serious conditions and that demonstrate the potential to address an unmet medical need. It was also granted priority review and orphan drug designation. Priority review status is granted to applications for drugs that, if approved, would be a significant improvement in safety or effectiveness in the treatment of a serious condition. Orphan drug designation provides incentives such as clinical trial tax credits, user fee waiver and eligibility for orphan drug exclusivity to assist and encourage the development of drugs for rare diseases.
The manufacturer received a rare pediatric disease priority review voucher, which comes from a program intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases. This is the seventh rare pediatric disease priority review voucher issued by the FDA since the program began.
Exondys 51 is made by Sarepta Therapeutics of Cambridge, Massachusetts.
Source: Sandy Walsh – FDA
Image Source: NeuroscienceNews.com image is credited to the CDC and is in the public domain.
[cbtabs][cbtab title=”MLA”]FDA “First Drug to Treat Duchenne Muscular Dystrophy Approved by FDA.” NeuroscienceNews. NeuroscienceNews, 19 September 2016.
<https://neurosciencenews.com/duchenne-muscular-dystrophy-drug-fda-5076/>.[/cbtab][cbtab title=”APA”]FDA (2016, September 19). First Drug to Treat Duchenne Muscular Dystrophy Approved by FDA. NeuroscienceNew. Retrieved September 19, 2016 from https://neurosciencenews.com/duchenne-muscular-dystrophy-drug-fda-5076/[/cbtab][cbtab title=”Chicago”]FDA “First Drug to Treat Duchenne Muscular Dystrophy Approved by FDA.” https://neurosciencenews.com/duchenne-muscular-dystrophy-drug-fda-5076/ (accessed September 19, 2016).[/cbtab][/cbtabs]