

Researchers at the RIKEN-Max Planck Joint Research Center in Japan have demonstrated that hallmark symptoms of Alzheimer’s disease can be reduced when sugars are prevented from binding to one of the key enzymes implicated in the disease. The new findings, reported in EMBO Molecular Medicine, show that abnormal attachment of a particular sugar to the enzyme BACE1 is a critical factor leading to the formation of Aβ plaques in the brain, and that plaques were reduced and cognitive performance improved when this action was prevented in mice through loss of the enzyme GnT-III. In doing so, this work has revealed a novel mechanism for Alzheimer’s disease development, potentially opening the way to a new approach for treatment.
A major factor contributing to Alzheimer’s disease is the formation of pathogenic Aβ plaques in the brain when the molecule APP is abnormally cut by the enzyme BACE1, producing Aβ. Because of this, developing drugs that prevent BACE1 from cleaving APP is a central focus of Alzheimer’s research. A major stumbling block in these efforts however, is finding a way to do this without disturbing vital processes in the brain and body that are regulated by normal BACE1 activity.
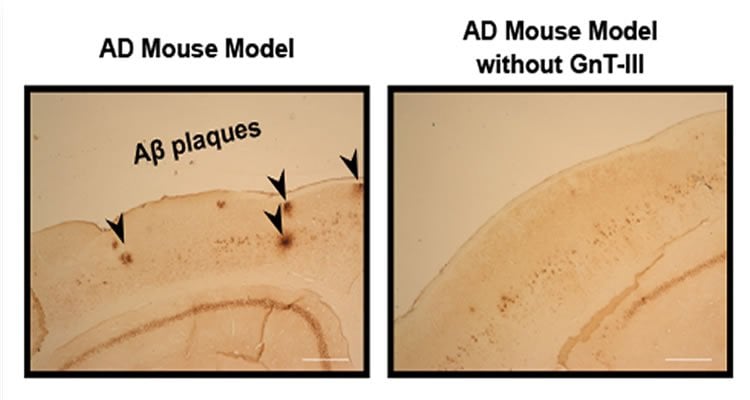
Through a series of experiments, researchers led by Yasuhiko Kizuka, Shinobu Kitazume, and Naoyuki Taniguchi at RIKEN, in collaboration with Tamao Endo and Shigeo Murayama at the Tokyo Metropolitan Institute of Gerontology, showed that much of the BACE1 found in the brains of Alzheimer’s disease patients is modified by the attachment of a particular sugar with the help of the enzyme GnT-III. Hypothesizing that preventing this process would relieve Alzheimer’s symptoms, the researchers crossed mice that lacked GnT-III with others that express human APP in the brain. They found that Aβ levels and plaque formation were reduced in these hybrid mice, and that cognitive performance was better.
Next, they showed that eliminating the sugared versions of BACE1 is effective in preventing plaque formation because BACE1 without the sugar is destroyed and cannot interact with APP. Using a difficult procedure called subcellular fractionation, they determined that sugared BACE1 is located near APP within cells. However, if the sugar is not attached to it, it is found further from APP and closer to lysosomes—subcellular structures that degrade unneeded molecules. When they examined the mice that lacked GnT-III, they indeed found lower amounts of BACE1.

They then showed that this approach is highly selective. BACE1 normally cleaves several proteins other than APP, and while mice lacking BACE1 also lack Aβ plaques, they also suffer from many side effects and often do not live longer than 4 weeks. On the other hand, the mice tested by Kizuka and his colleagues are healthy, fertile, and behave normally. When examined, these mice did not exhibit higher levels of other proteins that are normally cleaved by BACE1, showing that targeting GnT-III likely only prevents BACE1 from cleaving APP, and can act to reduce Alzheimer’s pathology with potentially few side effects.
This research highlights the importance of glycosylation—the modification of proteins by sugars. Far from being rare, it is the most common way in which protein structure is altered, with more than half of proteins being glycosylated in mammals. Kizuka is excited that perhaps now this process will get the attention and press it deserves. He notes that, “although a sugar change is often considered just a marker for disease or a specific cell type, our team has clearly demonstrated the functional role of a glycan during AD development.” He added that, “this work offers a good opportunity for many AD researchers to reconsider the importance of glycosylation.”
While knocking out GnT-III is feasible in mice, a drug that inhibits GnT-III in the brain has the potential to be an effective treatment for reducing Aβ plaque formation and cognitive decline in humans. Towards this end, Kizuka and colleagues are using a novel screening procedure in collaboration with the RIKEN Program for Drug discovery and Medical Technology Platforms to search for GnT-III inhibitors.
Contact: Naoyuki Taniguchi – RIKEN
Source: RIKEN press release
Image Source: The images are credited to Taniguchi et al. and are adapted from the RIKEN press release
Original Research: Full open access research for “An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease” by Yasuhiko Kizuka, Shinobu Kitazume, Reiko Fujinawa, Takashi Saito, Nobuhisa Iwata, Takaomi C. Saido, Miyako Nakano,Yoshiki Yamaguchi, Yasuhiro Hashimoto, Matthias Staufenbiel, Hiroyuki Hatsuta, Shigeo Murayama, Hiroshi Manya, Tamao Endo, and Naoyuki Taniguchi in EMBO Molecular Medicine. Published online January 15 2015 doi:10.15252/emmm.201404438
An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease
The β-site amyloid precursor protein cleaving enzyme-1 (BACE1), an essential protease for the generation of amyloid-β (Aβ) peptide, is a major drug target for Alzheimer’s disease (AD). However, there is a concern that inhibiting BACE1 could also affect several physiological functions. Here, we show that BACE1 is modified with bisecting N-acetylglucosamine (GlcNAc), a sugar modification highly expressed in brain, and demonstrate that AD patients have higher levels of bisecting GlcNAc on BACE1. Analysis of knockout mice lacking the biosynthetic enzyme for bisecting GlcNAc, GnT-III (Mgat3), revealed that cleavage of Aβ-precursor protein (APP) by BACE1 is reduced in these mice, resulting in a decrease in Aβ plaques and improved cognitive function. The lack of this modification directs BACE1 to late endosomes/lysosomes where it is less colocalized with APP, leading to accelerated lysosomal degradation. Notably, other BACE1 substrates, CHL1 and contactin-2, are normally cleaved in GnT-III-deficient mice, suggesting that the effect of bisecting GlcNAc on BACE1 is selective to APP. Considering that GnT-III-deficient mice remain healthy, GnT-III may be a novel and promising drug target for AD therapeutics.
“An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease” by Yasuhiko Kizuka, Shinobu Kitazume, Reiko Fujinawa, Takashi Saito, Nobuhisa Iwata, Takaomi C. Saido, Miyako Nakano,Yoshiki Yamaguchi, Yasuhiro Hashimoto, Matthias Staufenbiel, Hiroyuki Hatsuta, Shigeo Murayama, Hiroshi Manya, Tamao Endo, and Naoyuki Taniguchi in EMBO Molecular Medicine, doi:10.15252/emmm.201404438.




