Summary: By transplanting healthy glial cells, researchers have succeeded in reducing the symptoms and slowing the progression of Huntington’s disease in mice.
Source: University of Rochester Medical Center.
Researchers have successfully reduced the symptoms and slowed the progression of Huntington’s disease in mice using healthy human brain cells. The findings, which were published today in the journal Nature Communications, could ultimately point to a new method to treat the disease.
The research entailed implanting the animals with human glia cells derived from stem cells. One of the roles of glia, an important support cell found in the brain, is to tend to the health of neurons and the study’s findings show that replacing sick mouse glia with healthy human cells blunted the progress of the disease and rescued nerve cells at risk of death.
“The role that glia cells play in the progression of Huntington’s disease has never really been explored,” said Steve Goldman, M.D., Ph.D., co-director of the University of Rochester Center for Translational Neuromedicine. “This study shows that these cells are not only important actors in the disease, but may also hold the key to new treatment strategies.”
Huntington’s is a hereditary neurodegenerative disease that is most closely characterized by the loss of a specific nerve cell in the brain that plays a critical role in motor control called the medium spiny neurons. Over time, the disease results in involuntary movements, problems with coordination, and cognitive decline and depression. There is currently no way to slow or modify this fatal disease.
Most of the damage in Huntington’s disease occurs in a region of the brain called the striatum. Researchers have observed that as medium spiny neurons in the striatum die as a result of the disease, and that neighboring glial cells called astrocytes also become sick and do not function properly. However, it had not been clear if the sick astrocytes contributed to the signs and symptoms of the disease.
The researchers conducted a series of experiments in which they isolated human glial progenitors – the cells in the central nervous system that give rise to astrocytes – from both embryonic stem cells and brain tissue and implanted the cells into the striatum of mice with Huntington’s disease. Consistent with prior studies, they observed that the resulting human astrocytes outcompeted the native glia cells, resulting in mice with native neurons but human glia.
The researchers discovered that human glia transplanted into mice with the Huntington’s disease mutation appeared to keep neurons healthier and extended the animals survival. They also conducted a battery of tests designed to measure the animals’ behavior, memory, and motor skills, and the mice with healthy human glia performed significantly better than untreated mice with Huntington’s disease.
Conversely, when healthy mice were implanted with human glia carrying the genetic mutation that causes Huntington’s, the animals exhibited symptoms of the disease.
The researchers believe that the healthy human glia were able to essentially stabilize and perhaps even rescue neurons by restoring the normal signaling function that is lost during the disease. A complex series of chemical interactions must transpire when nerve cells fire and communicate with their neighbors. This activity requires neurons to constantly adjust and rebalance concentrations of important chemicals such as potassium, which participates in neuronal firing. Medium spiny neurons become overexcited in Huntington’s disease due to a genetic flaw that prevents potassium from entering the cells in sufficient amount – a condition that gives rise to the motor control and cognitive symptoms of the disease and produces a toxic chain reaction that ultimately kills the nerve cells.
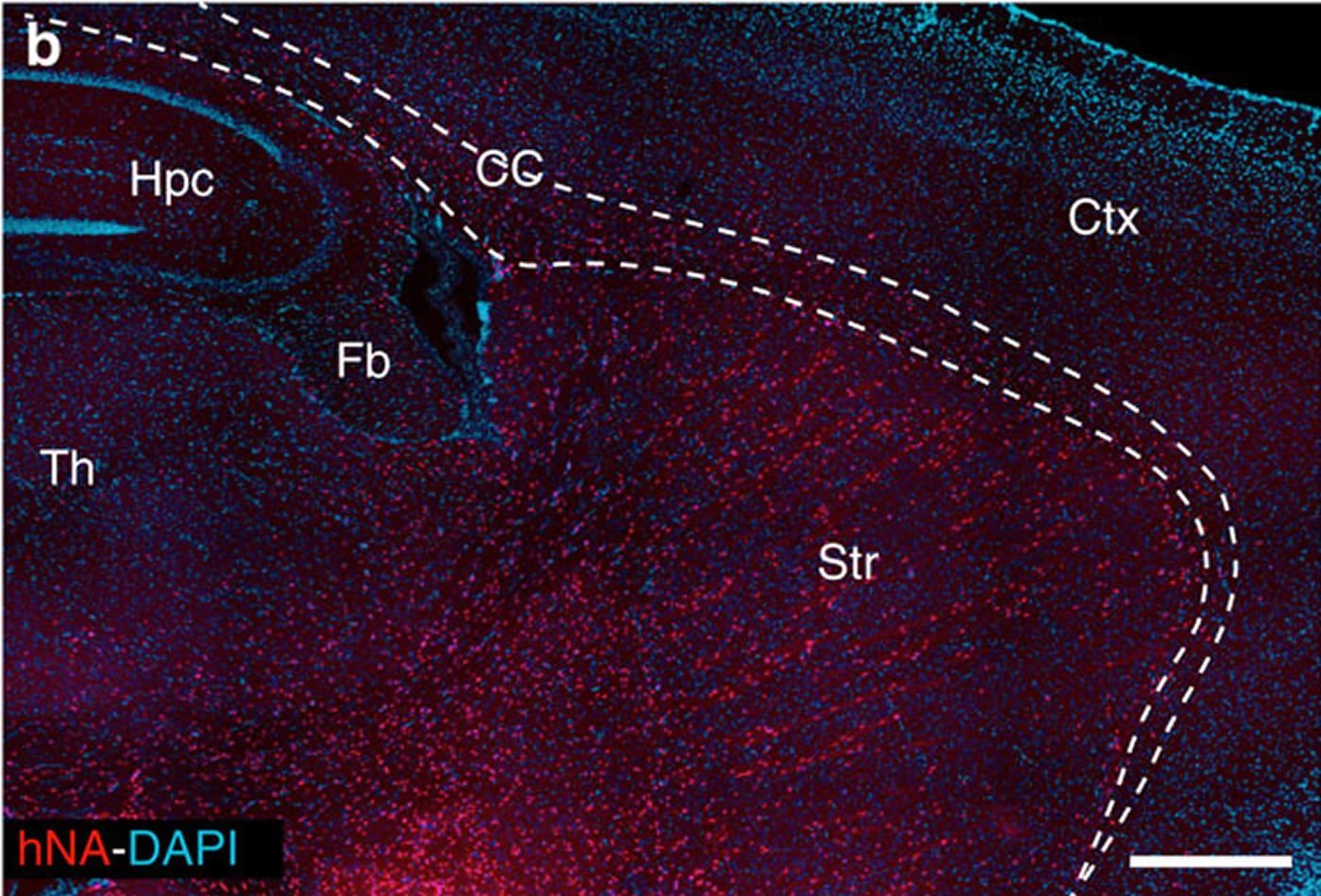
One of the roles of astrocytes is to function like a sponge and absorb potassium from the space surrounding neurons and create an environment that prevents neurons from becoming overactive. However, this function is impaired in glia in Huntington’s disease. The scientists found that the transplanted healthy glia were able to reestablish normal potassium uptake and thereby restore normal neuronal activity and rescue cells that might have otherwise died from hyper-excitability.
Because glia cells have been shown to migrate and proliferate throughout the brain once implanted, these findings could herald a potential new approach to rescue nerve cells threatened by the disease.
“The partial rescue of deficiencies we observed in this study tells us that there is a significant glia component in Huntington’s disease and that we may be able to improve function and delay progression with glial transplants,” said Goldman.
Additional co-authors of the study include Abdellatif Benraiss, Su Wang, Stephanie Herrlinger, Xialjie Li, Devin Chandler-Militello, Joseph Mauceri, Hayley Burm, Michael Toner, Qiwu Xu, Fengfei Ding, Fushun Wang, Ning Kang, Martha Windrem, and Maiken Nedergaard with the University of Rochester, Mikhail Osipovitch with the University of Copenhagen, Jian Kang with the New York Medical College, and Paul Curtin and Daniela Brunner with Psychogenetics, Inc. Goldman and Nedergaard maintain labs at both the University of Rochester and the University of Copenhagen.
Funding: The study was support with funds from the CHDI Foundation, the National Institutes of Health, the Leila Y. and G. Harold Mathers Charitable Foundation, the New York State Stem Cell Research Program, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation.
Source: Mark Michaud – University of Rochester Medical Center
Image Source: This NeuroscienceNews.com image is credited to the researcher/Nature Communications.
Original Research: Full open access research for “Human glia can both induce and rescue aspects of disease phenotype in Huntington disease” by Abdellatif Benraiss, Su Wang, Stephanie Herrlinger, Xiaojie Li, Devin Chandler-Militello, Joseph Mauceri, Hayley B. Burm, Michael Toner, Mikhail Osipovitch, Qiwu Jim Xu, Fengfei Ding, Fushun Wang, Ning Kang, Jian Kang, Paul C. Curtin, Daniela Brunner, Martha S. Windrem, Ignacio Munoz-Sanjuan, Maiken Nedergaard and Steven A. Goldman in Nature Communications. Published online June 7 2016 doi:10.1038/ncomms11758
[cbtabs][cbtab title=”MLA”]University of Rochester Medical Center. “Slowing Huntington’s by Swapping Sick for Healthy Brain Cells: Mouse Study.” NeuroscienceNews. NeuroscienceNews, 7 June 2016.
<https://neurosciencenews.com/huntingtons-stem-cell-transplant-4403/>.[/cbtab][cbtab title=”APA”]University of Rochester Medical Center. (2016, June 7). Slowing Huntington’s by Swapping Sick for Healthy Brain Cells: Mouse Study. NeuroscienceNews. Retrieved June 7, 2016 from https://neurosciencenews.com/huntingtons-stem-cell-transplant-4403/[/cbtab][cbtab title=”Chicago”]University of Rochester Medical Center. “Slowing Huntington’s by Swapping Sick for Healthy Brain Cells: Mouse Study.” https://neurosciencenews.com/huntingtons-stem-cell-transplant-4403/ (accessed June 7, 2016).[/cbtab][/cbtabs]
Abstract
Human glia can both induce and rescue aspects of disease phenotype in Huntington disease
The causal contribution of glial pathology to Huntington disease (HD) has not been heavily explored. To define the contribution of glia to HD, we established human HD glial chimeras by neonatally engrafting immunodeficient mice with mutant huntingtin (mHTT)-expressing human glial progenitor cells (hGPCs), derived from either human embryonic stem cells or mHTT-transduced fetal hGPCs. Here we show that mHTT glia can impart disease phenotype to normal mice, since mice engrafted intrastriatally with mHTT hGPCs exhibit worse motor performance than controls, and striatal neurons in mHTT glial chimeras are hyperexcitable. Conversely, normal glia can ameliorate disease phenotype in transgenic HD mice, as striatal transplantation of normal glia rescues aspects of electrophysiological and behavioural phenotype, restores interstitial potassium homeostasis, slows disease progression and extends survival in R6/2 HD mice. These observations suggest a causal role for glia in HD, and further suggest a cell-based strategy for disease amelioration in this disorder.
“Human glia can both induce and rescue aspects of disease phenotype in Huntington disease” by Abdellatif Benraiss, Su Wang, Stephanie Herrlinger, Xiaojie Li, Devin Chandler-Militello, Joseph Mauceri, Hayley B. Burm, Michael Toner, Mikhail Osipovitch, Qiwu Jim Xu, Fengfei Ding, Fushun Wang, Ning Kang, Jian Kang, Paul C. Curtin, Daniela Brunner, Martha S. Windrem, Ignacio Munoz-Sanjuan, Maiken Nedergaard and Steven A. Goldman in Nature Communications. Published online June 7 2016 doi:10.1038/ncomms11758