

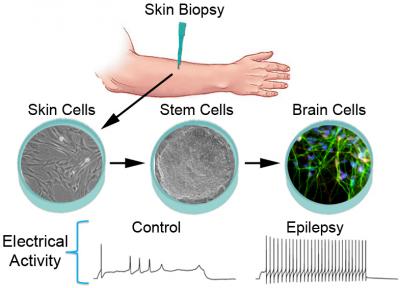
U-M-led study of neurons created from skin of patients with Dravet syndrome.
A new stem cell-based approach to studying epilepsy has yielded a surprising discovery about what causes one form of the disease, and may help in the search for better medicines to treat all kinds of seizure disorders.
The findings, reported by a team of scientists from the University of Michigan Medical School and colleagues, use a technique that could be called “epilepsy in a dish”.
By turning skin cells of epilepsy patients into stem cells, and then turning those stem cells into neurons, or brain nerve cells, the team created a miniature testing ground for epilepsy. They could even measure the signals that the cells were sending to one another, through tiny portals called sodium channels.
In neurons derived from the cells of children who have a severe, rare genetic form of epilepsy called Dravet syndrome, the researchers report abnormally high levels of sodium current activity. They saw spontaneous bursts of communication and “hyperexcitability” that could potentially set off seizures. Neurons made from the skin cells of people without epilepsy showed none of this abnormal activity.

They report their results online in the Annals of Neurology, and have further work in progress to create induced pluripotent stem cell lines from the cells of patients with other genetic forms of epilepsy. The work is funded by the National Institutes of Health, the American Epilepsy Society, the Epilepsy Foundation and U-M.
The new findings differs from what other scientists have seen in mice, demonstrating the importance of studying cells made from human epilepsy patients. Because the cells came from patients, they contained the hallmark seen in most patients with Dravet syndrome: a new mutation in SCN1A, the gene that encodes the crucial sodium channel protein called Nav1.1. That mutation reduces the number of channels to half the normal number in patients’ brains.
“With this technique, we can study cells that closely resemble the patient’s own brain cells, without doing a brain biopsy,” says senior author and team leader Jack M. Parent, M.D., professor of neurology at U-M and a researcher at the VA Ann Arbor Healthcare System. “It appears that the cells are overcompensating for the loss of channels due to the mutation. These patient-specific induced neurons hold great promise for modeling seizure disorders, and potentially screening medications.”
With the new paper, Parent, postdoctoral fellow Yu Liu, Ph.D. and their collaborators Lori Isom, Ph.D., professor of Pharmacology and of Molecular and Integrative Physiology at U-M, and Miriam Meisler, Ph.D., Distinguished University Professor of Human Genetics at U-M, report striking discoveries about what is happening at the cell level in the neurons of Dravet syndrome patients with a mutated SCN1A gene.
They also demonstrated that the effect is rooted in something that happens after function of the gene is reduced due to the mutation, though they don’t yet know how or why the nerve cells overcompensate for partial loss of this channel.
And, they found that the neurons didn’t show the telltale signs of hyperexcitability in the first few weeks after they were made — consistent with the fact that children with Dravet syndrome often don’t suffer their first seizures until they are several months old.
“In addition, reproduction of the hyperactivity of epileptic neurons in these cell cultures demonstrates that there is an intrinsic change in the neurons that does not depend on input from circuits in the brain,” says co-author Meisler.
A platform for testing medications
Many Dravet patients don’t respond to current epilepsy medications, making the search for new options urgent. Their lives are constantly under threat by the risk of SUDEP, sudden unexplained death in epilepsy – and they never outgrow their condition, which delays their development and often requires round-the-clock care.
“Working with patient families, and translating our sodium channel research to a pediatric disease, has made our basic science work much more immediate and critical,” says Isom, who serves on the scientific advisory board of the Dravet Syndrome Foundation along with Meisler. Parent, who co-directs U-M’s Comprehensive Epilepsy Program, was recently honored by the foundation.
The team is now working toward screening specific compounds for seizure-calming potential in Dravet syndrome, by testing their impact on the cells in the “epilepsy in a dish” model. The National Institutes of Health has made a library of drugs that have been approved by the U.S. Food and Drug Administration available for researchers to use — potentially allowing older drugs to have a second life treating an entirely different disease from what they were initially intended.
Parent and his colleagues hope to identify drugs that affect certain aspects of sodium channels, to see if they can dampen the sodium currents and calm hyperexcitability. The team is exploring new techniques that can make this process faster, using microelectrodes and calcium-sensitive dyes. They also hope to use the model to study potential drugs for non-genetic forms of epilepsy.
Having a U-M team that includes experts in induced pluripotent stem cell biology, sodium channel physiology and epilepsy genetics expertise helps the research progress, Parent notes. “Epilepsy is a complicated brain network disease,” he says. “It takes team-based science to address it.”
Patients as part of the research team
The U-M team’s research wouldn’t be possible without the participation of patients with Dravet syndrome and other genetic forms of epilepsy, and their parents.
More than 100 of them have joined the International Ion Channel Epilepsy Patient Registry, which is based at U-M and Miami Children’s Hospital and co-funded by the Dravet Syndrome Foundation and the ICE Epilepsy Alliance. The researchers hope to be able to conduct clinical trials of potential drugs with participation by these patients and others.
Meanwhile, patients with other genetically based neurological diseases can also help U-M scientists discover more about their conditions, by taking part in other efforts to create induced neurons from skin cells. Parent and his team have worked with several other U-M faculty to create stem cell lines from skin cells provided by patients with other diseases including forms of ataxia and lysosmal storage disease.
Notes about this neurology and epilepsy research
In addition to Parent, Liu, Meisler and Isom, the research team includes U-M’s Luis F. Lopez-Santiago, PhD, Yukun Yuan, PhD, Julie M. Jones, MS, Helen Zhang, MS, Heather A. O’Malley, Ph.D., Gustavo A. Patino, PhD, Janelle E. O’Brien, PhD, Raffaella Rusconi, PhD, and Robert C. Thompson, PhD, and the Cleveland Clinic’s Ajay Gupta, MD, and Marvin R. Natowicz, MD, PhD.
Funding: National Institute of Neurological Diseases and Stroke: RC1NS068684, NS064245, NS076752, National Heart, Lung and Blood Institute T32HL007853, National Institute of General Medical Sciences Genetics T32GM007544, U-M Rare Disease Initiative, Epilepsy Foundation, American Epilepsy Society, U-M Cardiovascular Center, Istituto Neurologico C. Besta, Milan, Italy.
Contact: Kara Gavin – University of Michigan Health System
Source: University of Michigan Health System press release
Image Source: The image is credited to Parent lab, University of Michigan Medical School and is adapted from the press release.
Original Research: Abstract for “Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism” by Yu Liu MD, PhD, Luis F. Lopez-Santiago PhD, Yukun Yuan PhD, Julie M. Jones MS, Helen Zhang MS, Heather A. O’Malley PhD, Gustavo A. Patino PhD, Janelle E. O’Brien PhD, Raffaella Rusconi PhD, Ajay Gupta MD, Robert C. Thompson PhD, Marvin R. Natowicz MD, PhD, Miriam H. Meisler PhD, Lori L. Isom PhD and Jack M. Parent MD in Annals of Neurology. Published online July 2 2013 DOI: 10.1002/ana.23897




