

Summary: Restoring HDAC1 could have potential benefits for those suffering from Alzheimer’s disease and people with age-related cognitive decline.
Source: MIT
MIT neuroscientists have discovered that an enzyme called HDAC1 is critical for repairing age-related DNA damage to genes involved in memory and other cognitive functions. This enzyme is often diminished in both Alzheimer’s patients and normally aging adults.
In a study of mice, the researchers showed that when HDAC1 is lost, a specific type of DNA damage builds up as the mice age. They also showed that they could reverse this damage and improve cognitive function with a drug that activates HDAC1.
The study suggests that restoring HDAC1 could have positive benefits for both Alzheimer’s patients and people who suffer from age-related cognitive decline, the researchers say.
“It seems that HDAC1 is really an anti-aging molecule,” says Li-Huei Tsai, the director of MIT’s Picower Institute for Learning and Memory and the senior author of the study. “I think this is a very broadly applicable basic biology finding, because nearly all of the human neurodegenerative diseases only happen during aging. I would speculate that activating HDAC1 is beneficial in many conditions.”
Picower Institute research scientist Ping-Chieh Pao is the lead author of the study, which appears today in Nature Communications.
DNA repair and aging
There are several members of the HDAC family of enzymes, and their primary function is to modify histones — proteins around which DNA is spooled. These modifications control gene expression by blocking genes in certain stretches of DNA from being copied into RNA.
In 2013, Tsai’s lab published two papers that linked HDAC1 to DNA repair in neurons. In the current paper, the researchers explored what happens when HDAC1-mediated repair fails to occur. To do that, they engineered mice in which they could knock out HDAC1 specifically in neurons and another type of brain cells called astrocytes.
For the first several months of the mice’s lives, there were no discernable differences in their DNA damage levels or behavior, compared to normal mice. However, as the mice aged, differences became more apparent. DNA damage began to accumulate in the HDAC1-deficient mice, and they also lost some of their ability to modulate synaptic plasticity — changes in the strength of the connections between neurons. The older mice lacking HCAC1 also showed impairments in tests of memory and spatial navigation.
The researchers found that HDAC1 loss led to a specific type of DNA damage called 8-oxo-guanine lesions, which are a signature of oxidative DNA damage. Studies of Alzheimer’s patients have also shown high levels of this type of DNA damage, which is often caused by accumulation of harmful metabolic byproducts. The brain’s ability to clear these byproducts often diminishes with age.
An enzyme called OGG1 is responsible for repairing this type of oxidative DNA damage, and the researchers found that HDAC1 is needed to activate OGG1. When HDAC1 is missing, OGG1 fails to turn on and DNA damage goes unrepaired. Many of the genes that the researchers found to be most susceptible to this type of damage encode ion channels, which are critical for the function of synapses.
Targeting neurodegeneration
Several years ago, Tsai and Stephen Haggarty of Harvard Medical School, who is also an author of the new study, screened libraries of small molecules in search of potential drug compounds that activate or inhibit members of the HDAC family. In the new paper, Tsai and Pao used one of these drugs, called exifone, to see if they could reverse the age-related DNA damage they saw in mice lacking HDAC1.
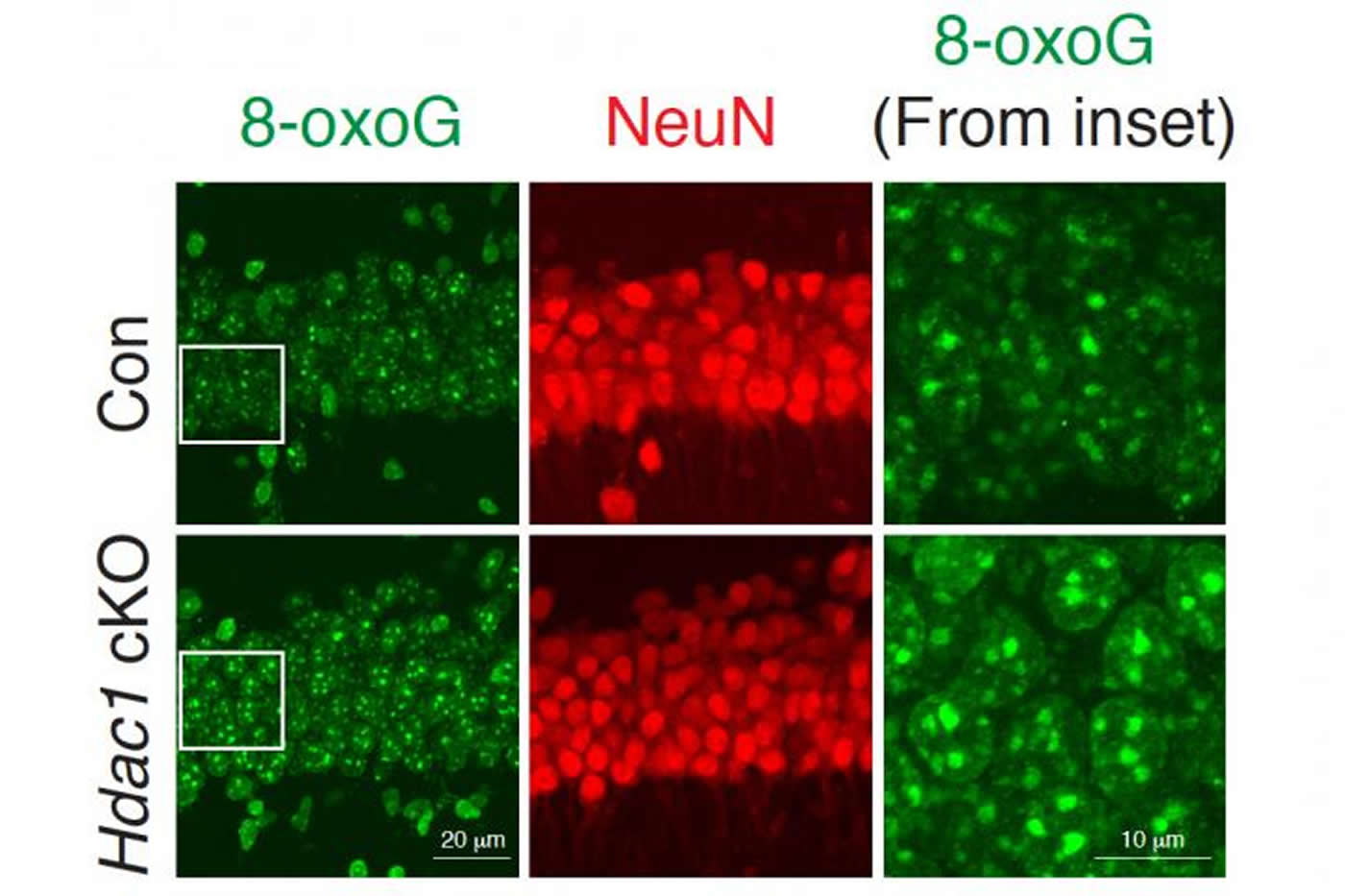
The researchers used exifone to treat two different mouse models of Alzheimer’s, as well as healthy older mice. In all cases, they found that the drug reduced the levels of oxidative DNA damage in the brain and improved the mice’s cognitive functions, including memory.
Exifone was approved in the 1980s in Europe to treat dementia but was later taken off the market because it caused liver damage in some patients. Tsai says she is optimistic that other, safer HDAC1-activating drugs could be worth pursuing as potential treatments for both age-related cognitive decline and Alzheimer’s disease.
“This study really positions HDAC1 as a potential new drug target for age-related phenotypes, as well as neurodegeneration-associated pathology and phenotypes,” she says.
Tsai’s lab is now exploring whether DNA damage and HDAC1 also play a role in the formation of Tau tangles — misfolded proteins in the brain that are a signature of Alzheimer’s and other neurodegenerative diseases.
Funding: The research was funded by the National Institute on Aging, the National Institute of Neurological Disorders and Stroke, and a Glenn Award for Research in Biological Mechanisms of Aging.
About this neuroscience research article
Source:
MIT
Media Contacts:
Sarah McDonnell – MIT
Image Source:
The image is credited to MIT.
Original Research: Open access
“HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease”. by Ping-Chieh Pao, Debasis Patnaik, L. Ashley Watson, Fan Gao, Ling Pan, Jun Wang, Chinnakkaruppan Adaikkan, Jay Penney, Hugh P. Cam, Wen-Chin Huang, Lorena Pantano, Audrey Lee, Alexi Nott, Trongha X. Phan, Elizabeta Gjoneska, Sara Elmsaouri, Stephen J. Haggarty & Li-Huei Tsai.
Nature Communications doi:10.1038/s41467-020-16361-y
Abstract
HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease
DNA damage contributes to brain aging and neurodegenerative diseases. However, the factors stimulating DNA repair to stave off functional decline remain obscure. We show that HDAC1 modulates OGG1-initated 8-oxoguanine (8-oxoG) repair in the brain. HDAC1-deficient mice display age-associated DNA damage accumulation and cognitive impairment. HDAC1 stimulates OGG1, a DNA glycosylase known to remove 8-oxoG lesions that are associated with transcriptional repression. HDAC1 deficiency causes impaired OGG1 activity, 8-oxoG accumulation at the promoters of genes critical for brain function, and transcriptional repression. Moreover, we observe elevated 8-oxoG along with reduced HDAC1 activity and downregulation of a similar gene set in the 5XFAD mouse model of Alzheimer’s disease. Notably, pharmacological activation of HDAC1 alleviates the deleterious effects of 8-oxoG in aged wild-type and 5XFAD mice. Our work uncovers important roles for HDAC1 in 8-oxoG repair and highlights the therapeutic potential of HDAC1 activation to counter functional decline in brain aging and neurodegeneration.
Feel Free To Share This Genetics News.




