

Deterioration of tiny brain region offers clues to onset of psychiatric disorder.
Scientists at Columbia University’s Mortimer B. Zuckerman Mind Brain Behavior Institute, Columbia University Medical Center (CUMC), and the Université Paris Descartes have found that deficits in social memory–a crucial yet poorly understood feature of psychiatric disorders such as schizophrenia–may be due to a decrease in the number of a particular class of brain cells, called inhibitory neurons, in a little-explored region within the brain’s memory center.
The findings, which were reported today in the journal Neuron, explain some of the underlying mechanisms that lead to the more difficult-to-treat symptoms of schizophrenia, including social withdrawal, reduced motivation and decreased emotional capacity.
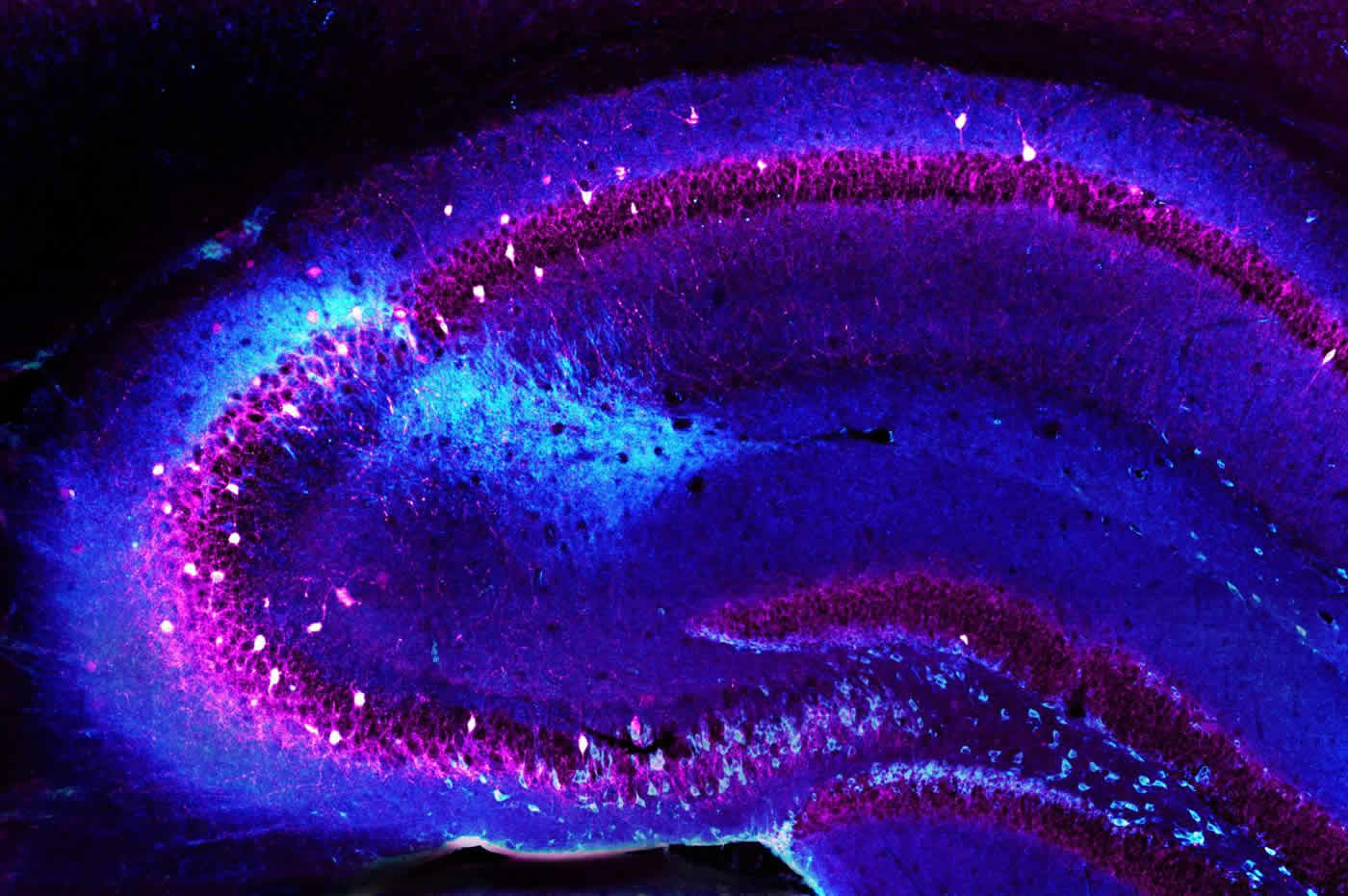
Scientists have long speculated that schizophrenia, which affects about 1 in every 100 adults worldwide, originates in part in the hippocampus–the brain’s headquarters for memory and spatial navigation. As a result, nearly every region of the hippocampus has been studied extensively in the hopes of gaining insight into the disorder. One notable exception is a tiny region of the hippocampus known as CA2.
“Smaller and less well-defined than other parts of the hippocampus, CA2 was like a small island that was depicted on old maps but remained unexplored,” explained Vivien Chevaleyre, PhD, group leader in neuroscience at the Université Paris Descartes and a lead author of the paper.
Several discoveries have focused attention on a possible association between CA2 and schizophrenia. This region of the hippocampus is associated with vasopressin, a hormone that plays a role in sexual bonding, motivation and other intensely social behaviors, which become impaired in people with the disorder. In addition, postmortem examinations of people with schizophrenia have revealed a marked decrease in the number of CA2 inhibitory neurons, while the rest of the hippocampus remained largely unaffected. However, the significance of this loss had remained unclear.
In this study, the researchers performed a series of electrophysiological and behavioral experiments on a mouse model of schizophrenia developed at CUMC.
By examining the brains of these mice, the researchers observed a substantial decrease in inhibitory CA2 neurons, as compared to a control group of normal, healthy mice–a change remarkably similar to that previously observed in postmortem examinations of people with schizophrenia. Moreover, the team discovered that the modified mice had a significantly reduced capacity for social memory compared with the controls. This raises the hypothesis that changes to CA2 may account for some of the social behavioral changes that occur in individuals with the disorder.
“Even the timing of the emergence of symptoms in the mice–during young adulthood–parallels the onset of schizophrenia in humans,” said Joseph Gogos, PhD, a professor of physiology and neuroscience at CUMC, a principal investigator at the Zuckerman Institute and a lead author of the paper.
“We can now examine the effects of schizophrenia at the cellular level and at the behavioral level,” said Steven Siegelbaum, PhD, chair of the Department of Neuroscience at CUMC, a principal investigator at the Zuckerman Institute and a co-author of the paper. “This essentially opens up a whole new avenue for research that could lead to earlier diagnosis and more effective treatments for schizophrenia.”
This research was supported by the CNRS ATIP-Avenir, Agence Nationale de la Recherche (ANR-12-BSV4-0021-01, ANR-13-JSV4-0002-01), the Ville de Paris and the National Institutes of Health (MH097879).
The authors report no financial or other conflicts of interest.
Source: Anne Holden – Zuckerman Mind Brain Behavior Institute
Image Credit: The image is credited to Vivien Chevaleyre
Original Research: Abstract for “Age-Dependent Specific Changes in Area CA2 of the Hippocampus and Social Memory Deficit in a Mouse Model of the 22q11.2 Deletion Syndrome” by Rebecca A. Piskorowski, Kaoutsar Nasrallah, Anastasia Diamantopoulou, Jun Mukai, Sami I. Hassan, Steven A. Siegelbaum, Joseph A. Gogos, and Vivien Chevaleyre in Neuron. Published online January 6 2016 doi:10.1016/j.neuron.2015.11.036
Abstract
Age-Dependent Specific Changes in Area CA2 of the Hippocampus and Social Memory Deficit in a Mouse Model of the 22q11.2 Deletion Syndrome
Highlights
•The 22q11.2 deletion mouse model displays unique changes in hippocampal area CA2
•There is an age-dependent reduction in PV+ interneurons and inhibitory transmission
•Inhibitory transmission is less plastic, and pyramidal cells in CA2 are less excitable
•Social memory is strongly impaired in the 22q11.2 deletion mouse model
Summary
Several neuropsychiatric disorders are associated with cognitive and social dysfunction. Postmortem studies of patients with schizophrenia have revealed specific changes in area CA2, a long-overlooked region of the hippocampus recently found to be critical for social memory formation. To examine how area CA2 is altered in psychiatric illness, we used the Df(16)A+/− mouse model of the 22q11.2 microdeletion, a genetic risk factor for developing several neuropsychiatric disorders, including schizophrenia. We report several age-dependent CA2 alterations: a decrease in the density of parvalbumin-expressing interneurons, a reduction in the amount of feedforward inhibition, and a change in CA2 pyramidal-neuron intrinsic properties. Furthermore, we found that area CA2 is less plastic in Df(16)A+/− mice, making it nearly impossible to evoke action potential firing in CA2 pyramidal neurons. Finally, we show that Df(16)A+/− mice display impaired social cognition, providing a potential mechanism and a neural substrate for this impairment in psychiatric disorders.
“Age-Dependent Specific Changes in Area CA2 of the Hippocampus and Social Memory Deficit in a Mouse Model of the 22q11.2 Deletion Syndrome” by Rebecca A. Piskorowski, Kaoutsar Nasrallah, Anastasia Diamantopoulou, Jun Mukai, Sami I. Hassan, Steven A. Siegelbaum, Joseph A. Gogos, and Vivien Chevaleyre in Neuron. Published online January 6 2016 doi:10.1016/j.neuron.2015.11.036




