Summary: Boosting mitochondria may defend against a form of protein stress that can reduce the formation of amyloid plaques, EPFL researchers report.
Source: EPFL.
Alzheimer’s disease is the most common form of dementia and neurodegeneration worldwide. A major hallmark of the disease is the accumulation of toxic plaques in the brain, formed by the abnormal aggregation of a protein called beta-amyloid inside neurons.
Still without cure, Alzheimer’s poses a significant burden on public health systems. Most treatments focus on reducing the formation of amyloid plaques, but these approaches have been inconclusive. As a result, scientists are now searching for alternative treatment strategies, one of which is to consider Alzheimer’s as a metabolic disease.
Taking this line of thought, Johan Auwerx’s lab at EPFL looked at mitochondria, which are the energy-producing powerhouses of cells, and thus central in metabolism. Using worms and mice as models, they discovered that boosting mitochondria defends against a particular form of protein stress, enables them to not only protect themselves, but to also reduce the formation of amyloid plaques.
During normal aging and age-associated diseases such as Alzheimer’s, cells face increasing damage and struggle to protect and replace dysfunctional mitochondria. Since mitochondria provide energy to brain cells, leaving them unprotected in Alzheimer’s disease favors brain damage, giving rise to symptoms like memory loss over the years.
The scientists identified two mechanisms that control the quality of mitochondria: First, the “mitochondrial unfolded protein response” (UPRmt), which protects mitochondria from stress stimuli. Second, mitophagy, a process that recycles defective mitochondria. Both these mechanisms are the key to delaying or preventing excessive mitochondrial damage during disease.
While we have known for a while that mitochondria are dysfunctional in the brains of Alzheimer’s patients, this is the first evidence that they actually try to fight the disease by boosting quality control pathways. “These defense and recycle pathways of the mitochondria are essential in organisms, from the worm C. elegans all the way to humans,” says Vincenzo Sorrentino, first author of the paper. “So we decided to pharmacologically activate them.”
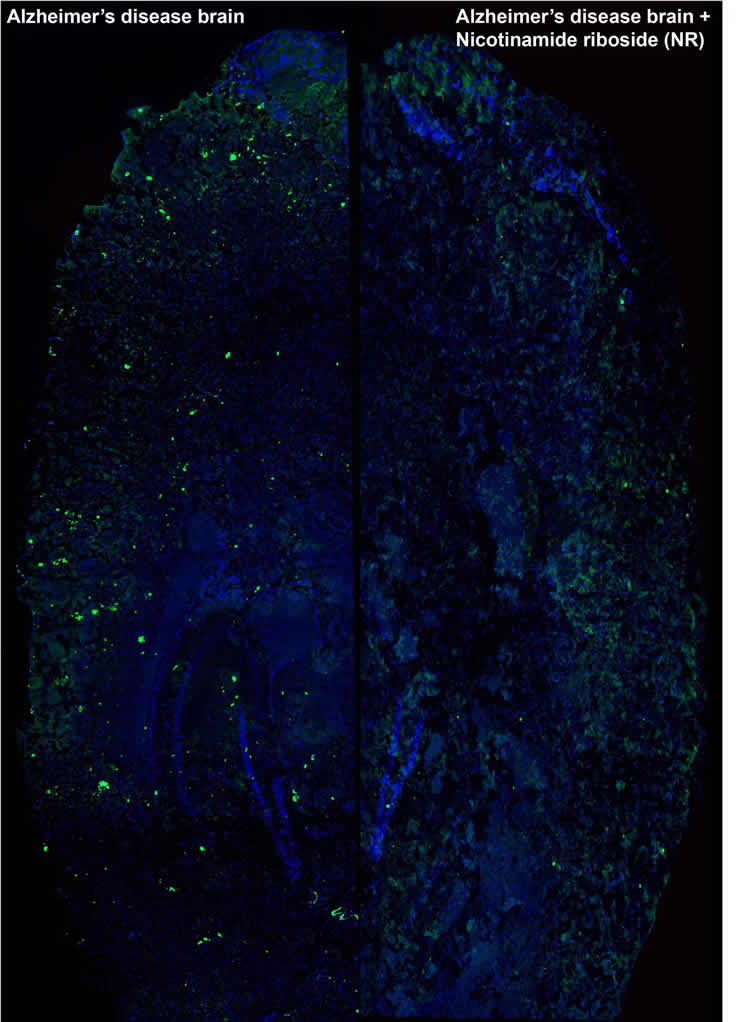
The team started by testing well-established compounds, such as the antibiotic doxycycline and the vitamin nicotinamide riboside (NR), which can turn on the UPRmt and mitophagy defense systems in a worm model (C. elegans) of Alzheimer’s disease. The health, performance and lifespan of worms exposed to the drugs increased remarkably compared with untreated worms. Plaque formation was also significantly reduced in the treated animals.
And most significantly, the scientists observed similar improvements when they turned on the same mitochondrial defense pathways in cultured human neuronal cells, using the same drugs.
The encouraging results led the researchers to test NR in a mouse model of Alzheimer’s disease. Just like C. elegans, the mice saw a significant improvement of mitochondrial function and a reduction in the number of amyloid plaques. But most importantly, the scientists observed a striking normalization of the cognitive function in the mice. This has tremendous implications from a clinical perspective.
According to Johan Auwerx, tackling Alzheimer’s through mitochondria could make all the difference. “So far, Alzheimer’s disease has been considered to be mostly the consequence of the accumulation of amyloid plaques in the brain,” he says. “We have shown that restoring mitochondrial health reduces plaque formation – but, above all, it also improves brain function, which is the ultimate objective of all Alzheimer’s researchers and patients.”
The strategy provides a novel therapeutic approach to slow down the progression of neurodegeneration in Alzheimer’s disease, and possibly even in other disorders such as Parkinson’s disease, which is also characterized by profound mitochondrial and metabolic defects.
The approach remains to be tested in human patients. “By targeting mitochondria, NR and other molecules that stimulate their ‘defense and recycle’ systems could perhaps succeed where so many drugs, most of which aim to decrease amyloid plaque formation, have failed,” says Vincenzo Sorrentino.
Researchers from Michigan State Uniersity also participated in this research.
Funding: Funding provided by EPFL Fellows’ Program, Associazione Italiana per la Ricerca sul Cancro, National Institutes of Health, Systems X, Velux Stiftung, Jebsen Foundation.
Source: Nik Papageorgiou – EPFL
Publisher: Organized by NeuroscienceNews.com.
Image Source: NeuroscienceNews.com image is credited to Vincenzo Sorrentino, Mario Romani, Francesca Potenza/EPFL.
Original Research: Abstract for “Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity” by Vincenzo Sorrentino, Mario Romani, Laurent Mouchiroud, John S. Beck, Hongbo Zhang, Davide D’ Amico, Norman Moullan, Francesca Potenza, Adrien W. Schmid, Solène Rietsch, Scott E. Counts & Johan Auwerx in Nature. Published online December 6 2017 doi:10.1038/nature25143
[cbtabs][cbtab title=”MLA”]EPFL “Healthy Mitochondria Could Stop Alzheimer’s.” NeuroscienceNews. NeuroscienceNews, 6 December 2017.
<https://neurosciencenews.com/alzheimers-mitochondria-8123/>.[/cbtab][cbtab title=”APA”]EPFL (2017, December 6). Healthy Mitochondria Could Stop Alzheimer’s. NeuroscienceNews. Retrieved December 6, 2017 from https://neurosciencenews.com/alzheimers-mitochondria-8123/[/cbtab][cbtab title=”Chicago”]EPFL “Healthy Mitochondria Could Stop Alzheimer’s.” https://neurosciencenews.com/alzheimers-mitochondria-8123/ (accessed December 6, 2017).[/cbtab][/cbtabs]
Abstract
Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity
Alzheimer’s disease is a common and devastating disease characterized by aggregation of the amyloid-β peptide. However, we know relatively little about the underlying molecular mechanisms or how to treat patients with Alzheimer’s disease. Here we provide bioinformatic and experimental evidence of a conserved mitochondrial stress response signature present in diseases involving amyloid-β proteotoxicity in human, mouse and Caenorhabditis elegans that involves the mitochondrial unfolded protein response and mitophagy pathways. Using a worm model of amyloid-β proteotoxicity, GMC101, we recapitulated mitochondrial features and confirmed that the induction of this mitochondrial stress response was essential for the maintenance of mitochondrial proteostasis and health. Notably, increasing mitochondrial proteostasis by pharmacologically and genetically targeting mitochondrial translation and mitophagy increases the fitness and lifespan of GMC101 worms and reduces amyloid aggregation in cells, worms and in transgenic mouse models of Alzheimer’s disease. Our data support the relevance of enhancing mitochondrial proteostasis to delay amyloid-β proteotoxic diseases, such as Alzheimer’s disease.
“Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity” by Vincenzo Sorrentino, Mario Romani, Laurent Mouchiroud, John S. Beck, Hongbo Zhang, Davide D’ Amico, Norman Moullan, Francesca Potenza, Adrien W. Schmid, Solène Rietsch, Scott E. Counts & Johan Auwerx in Nature. Published online December 6 2017 doi:10.1038/nature25143