Summary: Study reveals a self-corrective mechanism within synapses that is activated by neurodegeneration and slows disease progression in animal models of ALS.
Source: UCSF
A common feature of neurodegenerative diseases such as Alzheimer’s disease and amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease) is the progressive loss of synapses — the anatomical sites of communication between brain cells — throughout the brain and spinal cord. Typically, synapse loss becomes pervasive before the outward appearance of symptoms of disease, such as memory loss or paralysis. The fact that there must be extensive synapse loss before brain function begins to seriously decline suggests that the nervous system maintains a deep functional reserve that keeps everything working normally until the damage passes a tipping point and the brain’s resilience begins to break down.
But how exactly does this functional reserve confer resilience in the face of ongoing brain degeneration? Could differences in this reserve explain why some individuals with ALS decline and die within months, while others — like astrophysicist Steven Hawking — live for decades? And could a treatment that boosts this functional reserve help more patients survive and prosper as long as Hawking?
In a new study published May 6, 2020, in Neuron, UC San Francisco neuroscientist Graeme Davis, PhD, and his team have identified a powerful self-corrective mechanism within synapses that is activated by neurodegeneration and acts to slow down disease progression in animal models of ALS. Selectively eliminating this self-corrective mechanism dramatically accelerated progression of ALS in mice, shortening their lifespan by 50 percent.
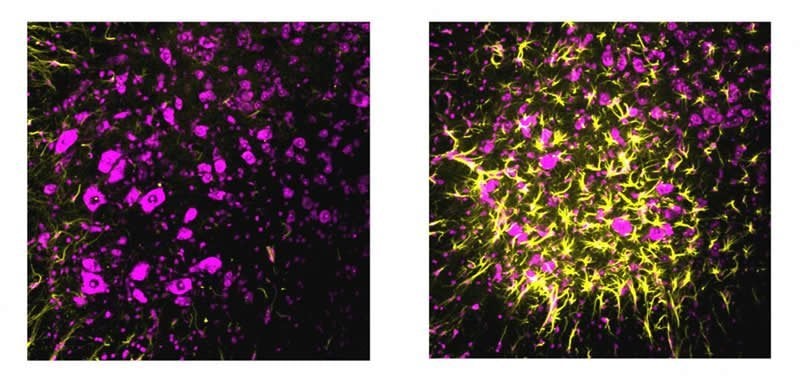
“Our data provide the first evidence that neurodegeneration kick-starts a self-corrective response that can keep the synapse between nerve and muscle working correctly, even though the disease process has already begun to nibble away at the synapse bit by bit, day by day. We also show for the first time that this self-corrective mechanism is amazingly potent — keeping mice with ALS alive twice as long as they would otherwise survive,” said Davis, the Morris Hertzstein Distinguished Professor of Medicine in the UCSF Department of Biochemistry and Biophysics and member of the UCSF Weill Institute for Neurosciences and UCSF Kavli Institute for Fundamental Neuroscience.
Davis has doggedly pursued the molecular underpinnings of self-corrective brain plasticity for decades, and his group is now translating their initial gene discovery efforts in mouse models of human diseases that include ALS, Alzheimer’s and epilepsy. Their goal is to leverage their most recent discoveries into a drug that boosts the brain’s self-corrective capacity, creating a greater functional reserve for people with ALS and other neurodegenerative diseases.
“Self-corrective plasticity could confer resilience to neurodegeneration of any kind and any cause, rather than being specific a single disease such as ALS,” Davis said. “I believe that we can improve the brain’s functional reserve, bringing everyone up to peak capacity and potentially extending lifespan across neurodegenerative diseases.”
Funding: The research was supported by the US National Institutes of Health (NIH) (NINDS R35NS097212, NIH HL146366, NHGRI R01HG003988, NHGRI 5K99HG009682) and the ALS Association (17-IIP-358), with research conducted at the E.O. Lawrence Berkeley National Laboratory and performed under Department of Energy Contract DE-AC02-05CH11231.
About this ALS research article
Source:
UCSF
Media Contacts:
Nicholas Weiler – UCSF
Image Source:
The image is credited to Davis Lab / UCSF.
Original Research: Closed access
“Presynaptic Homeostasis Opposes Disease Progression in Mouse Models of ALS-Like Degeneration: Evidence for Homeostatic Neuroprotection”. by Graeme Davis et al.
Neuron doi:10.1016/j.neuron.2020.04.009
Abstract
Presynaptic Homeostasis Opposes Disease Progression in Mouse Models of ALS-Like Degeneration: Evidence for Homeostatic Neuroprotection
Highlights
• In Drosophila and mouse, NMJ degeneration induces homeostatic plasticity
• Deletion of Scnn1a in motoneurons blocks homeostatic plasticity at the mouse NMJ
• Scnn1a cKO causes precocious disease progression in an ALS-like mutant background
• A model of “homeostatic neuroprotection” is proposed
Summary
Progressive synapse loss is an inevitable and insidious part of age-related neurodegenerative disease. Typically, synapse loss precedes symptoms of cognitive and motor decline. This suggests the existence of compensatory mechanisms that can temporarily counteract the effects of ongoing neurodegeneration. Here, we demonstrate that presynaptic homeostatic plasticity (PHP) is induced at degenerating neuromuscular junctions, mediated by an evolutionarily conserved activity of presynaptic ENaC channels in both Drosophila and mouse. To assess the consequence of eliminating PHP in a mouse model of ALS-like degeneration, we generated a motoneuron-specific deletion of Scnn1a, encoding the ENaC channel alpha subunit. We show that Scnn1a is essential for PHP without adversely affecting baseline neural function or lifespan. However, Scnn1a knockout in a degeneration-causing mutant background accelerated motoneuron loss and disease progression to twice the rate observed in littermate controls with intact PHP. We propose a model of neuroprotective homeostatic plasticity, extending organismal lifespan and health span.
Feel Free To Share This Neurology News.