Summary: A new study sheds light on the molecular mechanisms behind ALS.
Source: University of Montreal.
It took eight long years of research, but now an international team led by neuroscientists at Université de Montréal has discovered a basic molecular mechanism that better helps understand how Lou Gehrig’s disease, or amyotrophic lateral sclerosis (ALS), works.
And that basic science could someday lead to new therapy for the debilitating disease, which cripples people by removing the brain’s ability to communicate with their muscles, eventually leading to paralysis and early death.
“It’s a story of fundamental research about what happens normally in the body’s cells and what changes in the context of ALS,” said Jade-Emmanuelle Deshaies, a research associate in neurosciences at the UdeM Hospital Research Centre (CRCHUM) and lead author of the joint Canada-Israel study, published online today in the journal Brain.
“While studies such as this do not immediately give rise to new treatments for people living with ALS, they do deepen our understanding of the disease. ALS is very complicated; many cellular functions get mis-regulated. This type of work provides important information for future drug targets and the development of biomarkers aimed at detecting the disease more rapidly and following its progression.”
The research began eight years ago when Deshaies and her supervisor, associate professor of neurosciences Christine Vande Velde, started investigating what happens to various molecules when TDP-43, a protein that binds the ‘messengers’ in the cell known collectively as RNA and that is central to ALS pathology, is removed from the nucleus.
“Our specific focus was on other types of RNA binding proteins that could be interesting players in the disease,” Deshaies recalled. “One of these, hnRNP A1, caught our eye. In particular because there was a second form that is rarely mentioned in the literature.”
But first, a bit of basic science.
In molecular biology, genes encode RNA and the RNA then gets translated into proteins, the workhorses of cells. There are many different versions of RNA, each encoding many different versions of a protein. TDP-43, for one, binds RNA and can change how it is spliced – in a sequence of ABCD, for example, or of ABCEFG – a process called alternative splicing. Another RNA binding protein is hnRNP A1, and it gets spliced into two variants, both regulated by TDP-43.
Why is this important to understanding ALS? Because TDP-43 is known to be a major component of non-living substances in the cell called cytoplasmic inclusions, which are observed in 97 per cent of ALS cases.
“The data we have show that when TDP-43 is either not there at all, or is just absent from the nucleus, you can change the splicing pattern of hnRNP A1,” said Vande Velde. “The big picture is that there is a much more broad spectrum of RNA metabolism mis-regulation than what was previously thought. And with that, we get more understanding of what’s going wrong, and given this new knowledge, we can potentially develop a therapy that targets this mechanism.”
A parallel development is research into another motor neuron disease, spinal muscular atrophy (SMA). Scientists know that hnRNP A1 plays a role in its progress, controlling the splicing of an important gene called SMN, survival motor neuron. Vande Velde and her team don’t yet know whether or not the new splice variant they discovered changes SMN levels or function, but they point to a new drug therapy announced last year for SMA that does target the splicing of SMN by hnRNP A1.
“The drug is nusinersen, sold commercially as Spinraza,” Vande Velde explained. “When you give it to babies early enough, you can fix their spinal muscular atrophy. Babies that were not able to roll over, or walk, now can. Babies that would normally die within the first two or three years of life are able to reach the developmental milestones. It’s being reported as a real cure for the most severe forms of the disease.”
Developed by scientists at Cold Spring Harbor Laboratory and Ionis Pharmaceuticals, nusinersen is an exciting development “because they did the type of work we’ve been doing, which is to understand how a gene is spliced” said Vande Velde. That’s a therapeutic that came out of understanding a molecular mechanism involving alternative splicing.
“It took many years to get to that point, and similarly, our work is just the first cog in the wheel,” she added. “Whether or not there really is an influence on the expression or the splicing of the very important gene SMN, or other genes important for motor neuron survival, is something we need to evaluate,” Vande Velde said.
Her lab’s work is also a story of persistence. As Deshaies put it, “science is rarely straightforward. It often takes a winding road before leading to explanations and true understanding of what we observe.”
“We’re studying a mechanism that’s never been reported before,” Vande Velde noted. “We had some conflicting results early on, and it took time to figure out what the role of TDP-43 was in all this. It was “a massive team undertaking,” she added, “with team members from Israel (at Hebrew University), Quebec (at UdeM and Université de Sherbrooke) and elsewhere in Canada (at Western University) all making key contributions.”
More ALS research is done in Quebec than any other province, and the UdeM-led study was supported by grants from NSERC and the non-profit ALS Society of Canada. Hence the importance of getting the news out, even with no new therapy in sight.
“I think it’s an important service to communicate back to patients and their families what discoveries are being made with their donations, like those given via the Ice Bucket Challenge” said Vande Velde. “Many families are eager to learn about the molecular details of the disease and how we get to know them, and understand this is an important step in developing a therapy.”
Funding: Funding provided by Natural Sciences and Engineering Research Council, ALS Society of Canada.
Source: Jeff Heinrich – University of Montreal
Publisher: Organized by NeuroscienceNews.com.
Image Source: NeuroscienceNews.com image is credited to CRCHUM.
Original Research: Abstract in Brain.
doi:10.1093/brain/awy062
[cbtabs][cbtab title=”MLA”]University of Montreal “Better Understanding ALS by Looking at How Cells Change.” NeuroscienceNews. NeuroscienceNews, 19 March 2018.
<https://neurosciencenews.com/als-cell-changes-8662/>.[/cbtab][cbtab title=”APA”]University of Montreal (2018, March 19). Better Understanding ALS by Looking at How Cells Change. NeuroscienceNews. Retrieved March 19, 2018 from https://neurosciencenews.com/als-cell-changes-8662/[/cbtab][cbtab title=”Chicago”]University of Montreal “Better Understanding ALS by Looking at How Cells Change.” https://neurosciencenews.com/als-cell-changes-8662/ (accessed March 19, 2018).[/cbtab][/cbtabs]
Abstract
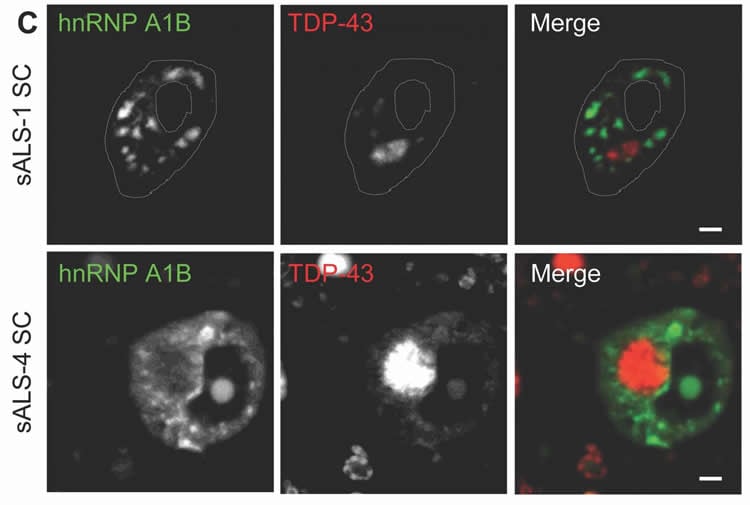
TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis
The RNA binding proteins TDP-43 (encoded by TARDBP) and hnRNP A1 (HNRNPA1) are each mutated in certain amyotrophic lateral sclerosis cases and are often mislocalized in cytoplasmic aggregates within motor neurons of affected patients. Cytoplasmic inclusions of TDP-43, which are accompanied by a depletion of nuclear TDP-43, are observed in most amyotrophic lateral sclerosis cases and nearly half of frontotemporal dementia cases. Here, we report that TDP-43 binds HNRNPA1 pre-mRNA and modulates its splicing, and that depletion of nuclear TDP-43 results in increased inclusion of a cassette exon in the HNRNPA1 transcript, and consequently elevated protein levels of an isoform containing an elongated prion-like domain, referred to as hnRNP A1B. Combined in vivo and in vitro approaches demonstrated greater fibrillization propensity for hnRNP A1B, which drives protein aggregation and is toxic to cells. Moreover, amyotrophic lateral sclerosis patients with documented TDP-43 pathology showed neuronal hnRNP A1B cytoplasmic accumulation, indicating that TDP-43 mislocalization may contribute to neuronal vulnerability and loss via altered HNRNPA1 pre-mRNA splicing and function. Given that TDP-43 and hnRNP A1 each bind, and thus modulate, a third of the transcriptome, our data suggest a much broader disruption in RNA metabolism than previously considered.