

Using the genetic information of two different families with three generations of disease, researchers have identified a new mutation responsible for a degenerative and ultimately fatal movement disorder. Through induced pluripotent stem cell techniques, researchers also grew neurons from one patient in the laboratory to be used in future experiments.
Spinocerebellar ataxia (SCA) is a genetic disease that causes wasting away of the cerebellum, the portion of the brain responsible for controlling voluntary muscle movement, like walking, speaking, and even the direction of our eyes.
Currently, SCA has no cure or treatment. The mutations responsible for about 30 percent of cases are still unidentified.
Two different families with SCA sought treatment at two different hospitals in Japan. After preliminary testing on the symptomatic individuals, doctors identified none of the known genetic mutations. Researchers at Hiroshima University then received the patient’s genetic samples and began the process of searching for the new mutation.
After genetic sequencing of four family members with SCA, a research team led by Professor Hideshi Kawakami, MD, PhD, from the Department of Epidemiology at Hiroshima University used statistical analysis to compare the families’ DNA to that of unrelated people without SCA. This statistical analysis allowed researchers to identify which genetic variation the family members with SCA shared that healthy people did not.
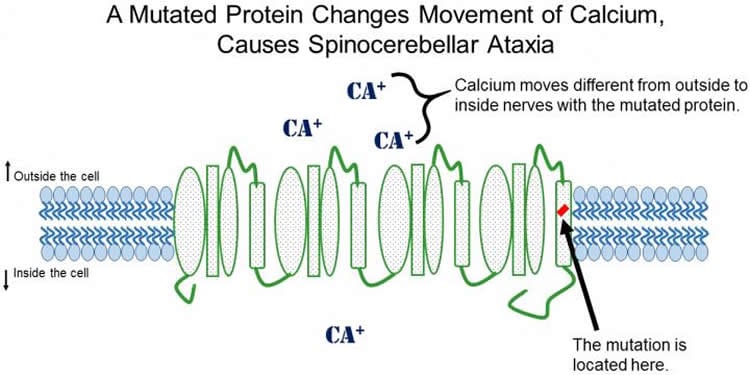
The gene responsible for causing both families’ SCA is located on Chromosome 17. The gene, called CACNA1G, encodes the Cav3.1 protein. Cav3.1 serves as a type of ion channel, or gateway, between the inside of nerve cells and the rest of the body. Scientists in different fields of research already know Cav3.1 controls how many Calcium ions are allowed into nerves when they send an electrical impulse through the brain. Cav3.1 had never been linked to SCA before.
Changing a single letter in the DNA sequence of CACNA1G switches a single amino acid in the chain of 2377 amino acids that cells connect to build the Cav3.1 protein.
Researchers performed the experiments to examine the way the mutated Cav3.1 channel behaves in cells growing in a dish. This mutation makes the Cav3.1 channels open at a lower threshold, meaning they let Calcium into the cell differently from healthy cells.
“In the future, a drug modifying this channel may cure the patients,” said Prof. Kawakami.
Skin cells from one patient were used in induced pluripotent stem cell experiments to grow this patient’s neurons in the laboratory. These new neurons showed no obvious physical deformities, which might fit with normal SCA progression. Depending on which SCA mutation they have, some patients may not experience symptoms until middle-age.
“We might need some age-related factors to reproduce life-like cell behavior,” said Prof. Kawakami.
Researchers plan to use the neurons in future experiments to study the disease-causing Cav3.1 under more life-like conditions and in greater detail.
Funding: Funding was provided by Ministry of Education, Culture, Sports, Science and Technology of Japan, Japan Society for the Promotion of Science, Core Program for Disease Modeling Using iPS Cells from AMED.
Source: Norifumi Miyokawa – Hiroshima University
Image Source: The image is credited to Hiroshima University.
Original Research: Full open access research for “A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia” to Hiroyuki Morino, Yukiko Matsuda, Keiko Muguruma, Ryosuke Miyamoto, Ryosuke Ohsawa, Toshiyuki Ohtake, Reiko Otobe, Masahiko Watanabe, Hirofumi Maruyama, Kouichi Hashimoto and Hideshi Kawakami in Molecular Brain. Published online December 29 2015 doi:10.1186/s13041-015-0180-4
Abstract
A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia
Background
Spinocerebellar ataxia (SCA) is a genetically heterogeneous disease. To date, 36 dominantly inherited loci have been reported, and 31 causative genes have been identified.
Results
In this study, we analyzed a Japanese family with autosomal dominant SCA using linkage analysis and exome sequencing, and identified CACNA1G, which encodes the calcium channel CaV3.1, as a new causative gene. The same mutation was also found in another family with SCA. Although most patients exhibited the pure form of cerebellar ataxia, two patients showed prominent resting tremor in addition to ataxia. CaV3.1 is classified as a low-threshold voltage-dependent calcium channel (T-type) and is expressed abundantly in the central nervous system, including the cerebellum. The mutation p.Arg1715His, identified in this study, was found to be located at S4 of repeat IV, the voltage sensor of the CaV3.1. Electrophysiological analyses revealed that the membrane potential dependency of the mutant CaV3.1 transfected into HEK293T cells shifted toward a positive potential. We established induced pluripotent stem cells (iPSCs) from fibroblasts of the patient, and to our knowledge, this is the first report of successful differentiation from the patient-derived iPSCs into Purkinje cells. There was no significant difference in the differentiation status between control- and patient-derived iPSCs.
Conclusions
To date, several channel genes have been reported as causative genes for SCA. Our findings provide important insights into the pathogenesis of SCA as a channelopathy.
“A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia” to Hiroyuki Morino, Yukiko Matsuda, Keiko Muguruma, Ryosuke Miyamoto, Ryosuke Ohsawa, Toshiyuki Ohtake, Reiko Otobe, Masahiko Watanabe, Hirofumi Maruyama, Kouichi Hashimoto and Hideshi Kawakami in Molecular Brain. Published online December 29 2015 doi:10.1186/s13041-015-0180-4




