

Highlighting a potential target in the treatment of multiple sclerosis (MS) and Alzheimer’s disease, new research suggests that triggering a protein found on the surface of brain cells may help slow the progression of these and other neurological diseases.
Working with mice, two research teams at Washington University School of Medicine in St. Louis independently linked the protein to the ability to clear debris from the brain. Such waste builds up both as a byproduct of daily mental activities and as a result of misdirected immune system attacks on brain cells. If too much debris is present in the brain for too long, it can contribute to neurological disease.
In one study, appearing online Feb. 26 in Cell, scientists showed that Alzheimer’s brain plaques build up more slowly in mice that have a defective version of the TREM2 protein. In another, published Jan. 29 in Acta Neuropathologica, researchers showed that mice lacking the same protein had trouble cleaning up debris in the brain produced by damage to a protective coating on nerve cells. The problem is thought to occur in MS and other neurological disorders.
“We’ve been very interested in identifying ways to control naturally occurring mechanisms that help clean and repair the brain, and these new studies provide clear evidence that TREM2 could be just such a target,” said Laura Piccio, MD, PhD assistant professor of neurology and senior author of one of the studies.
Scientists are looking for ways to activate the protein to slow or prevent damage caused by neurological disorders.
Previous studies have linked rare forms of the TREM2 gene to early-onset dementia and increased risk of Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis (ALS).
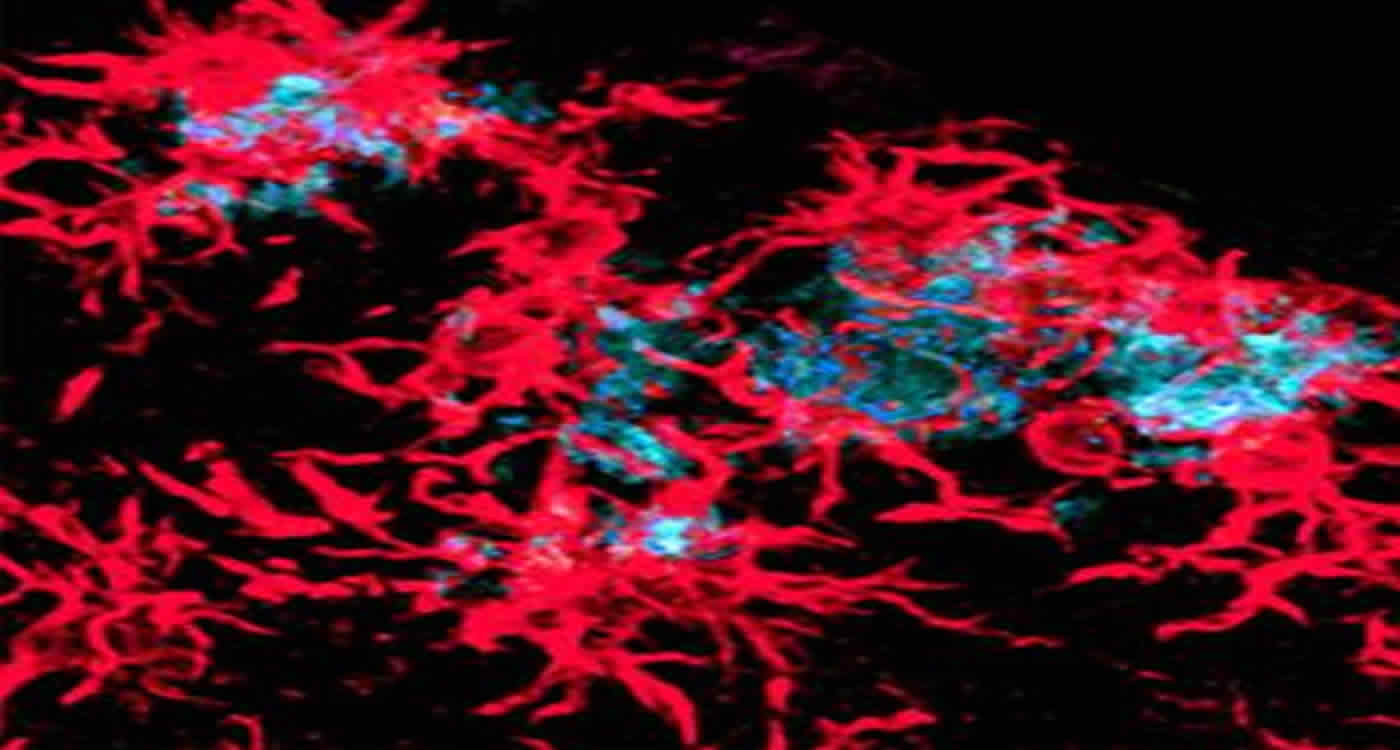
Scientists knew the protein was found on brain cells called microglia, which help maintain and repair the central nervous system. The new studies are among the first to provide clear evidence that the protein plays an integral role in at least some of these processes.
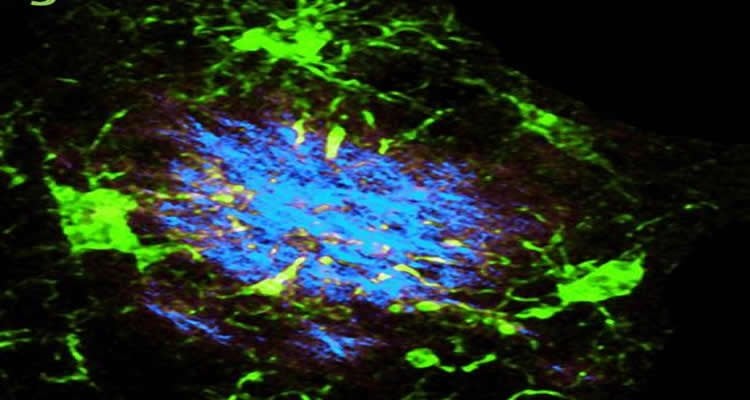
In Alzheimer’s disease, amyloid beta, a byproduct of brain metabolism that is normally cleared from the brain, builds up to form plaques. Researchers in the laboratories of Marco Colonna, MD, the Robert Rock Belliveau MD Professor of Pathology, and John Cirrito, PhD, associate professor of neurology, bred mice lacking the gene with mice genetically engineered to have an Alzheimer’s-like condition.
First author Yaming Wang, PhD, a postdoctoral research scholar, monitored the buildup of amyloid plaques in the mice offspring as they aged and found that the absence of the gene significantly accelerated the accumulation of the plaques.
“We found that microglia cluster around amyloid plaques when TREM2 is present, presumably because the cells are getting ready to absorb the plaques and break them down,” said Colonna. “When TREM2 is absent, this clustering does not occur.”
In MS, misdirected immune cell attacks damage myelin, a protective coating on nerve cells, leaving myelin fragments in brain tissue. Failure to promptly remove this debris can worsen damage caused by the condition and inhibit repair mechanisms.
For the MS study, Piccio and colleagues at the John L. Trotter MS Center at Washington University School of Medicine and Barnes-Jewish Hospital gave a compound called cuprizone to mice that lacked the TREM2 gene. Cuprizone causes loss of myelin in a manner somewhat similar to that seen in people with MS.
“When we give normal mice this chemical, they can clear most of the myelin fragments from the brain,” Piccio said. “But when we gave cuprizone to mice that did not have the gene and looked at their brains four, six and 12 weeks later, we could still see evidence of damaged myelin.”
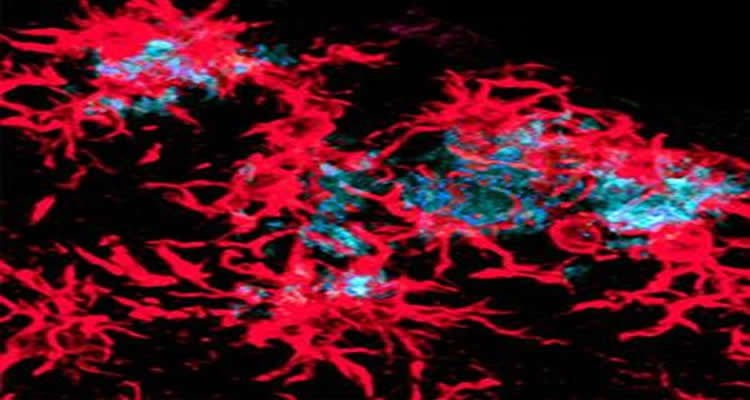
Motor coordination in these mice also was significantly more impaired after cuprizone exposure. This may reflect enhanced damage to brain cells resulting from the lingering presence of damaged myelin in the brain.
Colonna and his colleagues showed that TREM2 detects molecules associated with amyloid beta and with damaged neurons. They believe that the protein helps keep microglia from self-destructing as debris is cleared from the brain.
“This is a mechanism that is very common in immune cells,” he explained. “When a signal activates immune cells and they start attacking an invader or working to repair an injury, they start using energy very rapidly. If the cells do not receive a second signal confirming the need for their services, this increased energy usage will kill them.”
The Cell research was supported by the Lilly Innovation Fellowship Award; the Washington University Digestive Diseases Research Core Center, grant P30 DK52574; the National Multiple Sclerosis Society; the Knight Alzheimer’s Disease Research Center, pilot grant P50 AG005681-30; and the Cure Alzheimer’s Fund.
The Acta Neuropathologica research was supported by the Fondazione Italiana Sclerosi Multipla, the National Multiple Sclerosis Society and the Dana Foundation.
Contact: Michael C. Purdy – Washington University School of Medicine
Source: Washington University School of Medicine press release
Image Source: The images are credited to Yaming Wang/Bernd Zinselmeyer and are adapted from the Washington University School of Medicine press release
Original Research: Abstract for “TREM2 lipid sensing sustain microglial response in an Alzheimer’s disease model” by Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, and Colonna M. in Cell. Published online February 26 2015, doi:10.1016/j.cell.2015.01.049
Abstract for “TREM2 regulates microglial cell activation in response to demyelination in vivo” by Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, Yuede CM, Galimberti D, Olivecrona G, Klein RS, Cross AH, and Otero K, Piccio L. in Acta Neuropathologica. Published online January 29 2015 DOI:10.1007/s00401-015-1388-1




