Summary: Over-expression of the synaptic protein RAPGEF2 drives synaptic loss associated with Alzheimer’s disease.
Source: Korea Brain Research Institute
Korea Brain Research Institute (KBRI, Pann-Ghill Suh (President)) announced that Dr. Kea Joo Lee and Dr. You-Na Jang of the Neural Circuits Research Group have identified the mechanism causing synaptic loss in Alzheimer’s disease as the aberrant expression of RAPGEF2, a synaptic protein.
The results were published on January 2021, in the online Early View of Neuropathology and Applied Neurobiology.
Alzheimer’s disease (AD) accounts for about 75% of dementia cases and is the most common type of degenerative brain disease. AD is a devastating because disease progression can cause memory loss, mood disorder, slurred speech, confusion, and impaired movement.
With the conventional treatment available today, AD patients may expect to see some temporary improvement of symptoms, but nothing exists at the moment that can halt or reverse the progression. Instead, preventive strategies are emphasized, with health care professionals commonly suggesting physical exercise and continued learning programs as options.
AD is a tricky condition that has constantly thwarted the best efforts to unravel the inner workings of the disease. In the leading hypothesis, abnormal aggregation of amyloid beta (Aß) and tau proteins are identified as a possible cause of the illness. Amyloid beta is known to degrade synapses and drive cognitive impairment such as memory loss.
In their work unveiling the complex processes by which amyloid beta brings about synaptic loss, Dr. Kea Joo Lee and his team have been studying the brain tissue of both deceased Alzheimer’s patients and genetically modified mouse models for the disease, and have found “RAPGEF2 protein overexpression” to be the common phenomenon.
RAPGEF2(Rap guanin nucleotide exchange factor 2) is an essential protein involved in multiple critical biological pathways such as synaptic remodeling, neural plasticity, and embryo neural development
Employing various neurobiological methodologies that utilize neuronal cell culture models and brain tissue from mouse models of Alzheimer’s, the researchers arrived at the conclusion that “amyloid beta facilitates the overexpression of the RAPGEF2 protein,” and “the RAPGEF2 protein, in turn, activates downstream effectors RAP2 and JNK to ultimately induce synaptic loss.”
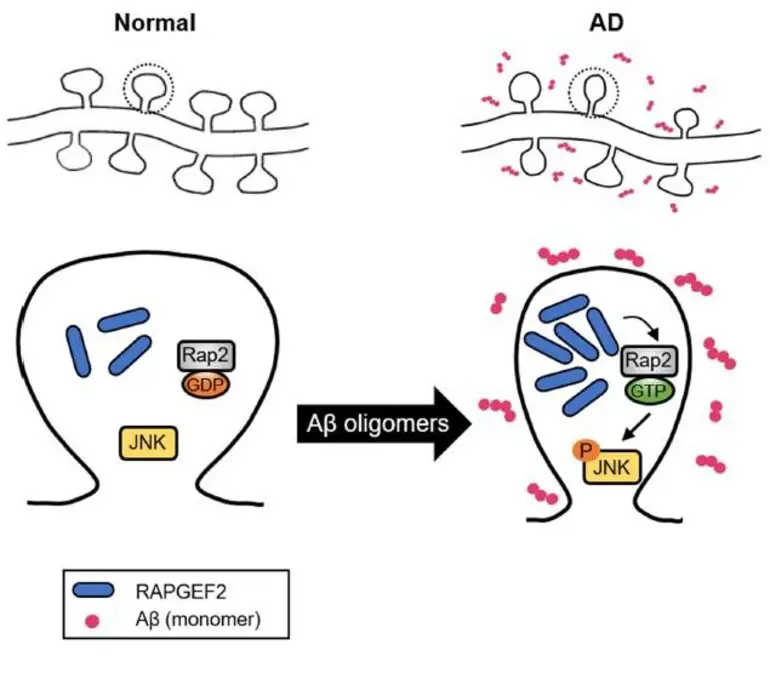
Intriguingly, electron microscopy and behavioral tests conducted by the team showed the silencing of RAPGEF2 as having a preventive effect on synapse loss and cognitive impairment even in the presence of increased amyloid beta.
The potential significance of these findings is great. Having a detailed understanding of the molecular mechanisms behind synaptic damage that occurs in the early stages of the Alzheimer’s disease can be invaluable in developing treatments for neurodegenerative diseases such as dementia which have continued to plague humanity despite scientific advancement.
Funding: This research was supported by grants from the National Research Foundation of Korea, and the Korea Brain Research Institute (KBRI) basic research program.
About this Alzheimer’s disease research news
Source: Korea Brain Research Institute
Contact: Kea Joo Lee – Korea Brain Research Institute
Image: The image is credited to Korea Brain Research Institute
Original Research: Open access.
“RAPGEF2 mediates oligomeric Aβ‐induced synaptic loss and cognitive dysfunction in the 3xTg‐AD mouse model of Alzheimer’s disease” by Kea Joo Lee et al. Neuropathology and Applied Neurobiology
Abstract
RAPGEF2 mediates oligomeric Aβ‐induced synaptic loss and cognitive dysfunction in the 3xTg‐AD mouse model of Alzheimer’s disease
Aims
Amyloid‐β (Aβ) oligomers trigger synaptic degeneration that precedes plaque and tangle pathology. However, the signalling molecules that link Aβ oligomers to synaptic pathology remain unclear. Here, we addressed the potential role of RAPGEF2 as a novel signalling molecule in Aβ oligomer‐induced synaptic and cognitive impairments in human‐mutant amyloid precursor protein (APP) mouse models of Alzheimer’s disease (AD).
Methods
To investigate the role of RAPGEF2 in Aβ oligomer‐induced synaptic and cognitive impairments, we utilised a combination of approaches including biochemistry, molecular cell biology, light and electron microscopy, behavioural tests with primary neuron cultures, multiple AD mouse models and post‐mortem human AD brain tissue.
Results
We found significantly elevated RAPGEF2 levels in the post‐mortem human AD hippocampus. RAPGEF2 levels also increased in the transgenic AD mouse models, generating high levels of Aβ oligomers before exhibiting synaptic and cognitive impairment. RAPGEF2 upregulation activated the downstream effectors Rap2 and JNK. In cultured hippocampal neurons, oligomeric Aβ treatment increased the fluorescence intensity of RAPGEF2 and reduced the number of dendritic spines and the intensities of synaptic marker proteins, while silencing RAPGEF2 expression blocked Aβ oligomer‐induced synapse loss. Additionally, the in vivo knockdown of RAPGEF2 expression in the AD hippocampus prevented cognitive deficits and the loss of excitatory synapses.
Conclusions
These findings demonstrate that the upregulation of RAPGEF2 levels mediates Aβ oligomer‐induced synaptic and cognitive disturbances in the AD hippocampus. We propose that an early intervention regarding RAPGEF2 expression may have beneficial effects on early synaptic pathology and memory loss in AD.