

Summary: Researchers have developed a neurodevelopmental model of Williams syndrome that may provide insights into the neurobiology of the social brain.
Source: UCSD.
Rare genetic condition produces individuals with extremely sociable personalities but research may also shed light on biology and behavior of persons with autism and other social disorders.
In a study spanning molecular genetics, stem cells and the sciences of both brain and behavior, researchers at University of California San Diego, with colleagues at the Salk Institute of Biological Studies and elsewhere, have created a neurodevelopmental model of a rare genetic disorder that may provide new insights into the underlying neurobiology of the human social brain.
The findings are published in the August 10 online edition of Nature.
Scientists investigated Williams syndrome or WS, a rare genetic condition caused by deletion of one copy of 25 contiguous genes on chromosome 7, out of an estimated 30,000 genes in the brain. WS affects one in 10,000 people worldwide, and an estimated 20,000 Americans. The condition occurs equally in both genders and across cultures.
WS results in a host of medical problems as well as a specific heart defect. Persons with the deletion typically display a distinctive face with a small, upturned nose, wide mouth, full lips and small chin and may also have dental and orthopedic problems. Neurologically, they have developmental delays, with severe spatial deficits, yet relative strengths in language use and face processing.
“An interesting aspect is the typical hyper-social predisposition,” said study co-author Ursula Bellugi, EdD, director of the cognitive neuroscience lab at Salk and an adjunct professor at UC San Diego who has studied WS for years. “Persons with the WS deletion tend to be overly friendly, overly trusting, drawn to strangers, yet anxious.”
But Bellugi said it has not been clear how genetics links to the behavioral aspects of WS. “A human model for the disease could fill in the scientific gaps and would help to understand the mechanisms behind the disorder. WS is an elegant model for being able to go across levels,” she said.
Co-senior study author Alysson Muotri, PhD, associate professor of pediatrics and cellular and molecular medicine at UC San Diego School of Medicine, became intrigued by WS because the condition is so different from his usual research focus on autism, which is characterized by lower sociability and language skills.
“I was fascinated on how a genetic defect, a tiny deletion in one of our chromosomes, could make us friendlier, more empathetic and more able to embrace our differences,” Muotri said.
In recent years, Muotri and colleagues have created in vitro cellular models of autism using reprogrammed induced pluripotent stem cells (iPSC) derived from discarded baby teeth of children with autism, work dubbed the “tooth fairy project.” They did so again here.
The team began with dental pulp cells extracted from teeth donated by young children with WS. The cells were reprogrammed to become neural progenitor cells able to form functional neuronal networks resembling the developing cortex of the human brain in a dish.
“We discovered that WS neural progenitor cells failed to proliferate due to high levels of cell death,” said Muotri. “And as a consequence of the lower replication of progenitor cells, WS brains have reduced cortex surface area.” The observation was validated using magnetic resonance imaging of live study participants by Eric Halgren, PhD, professor in the Department of Neurosciences at UC San Diego School of Medicine, and colleagues.
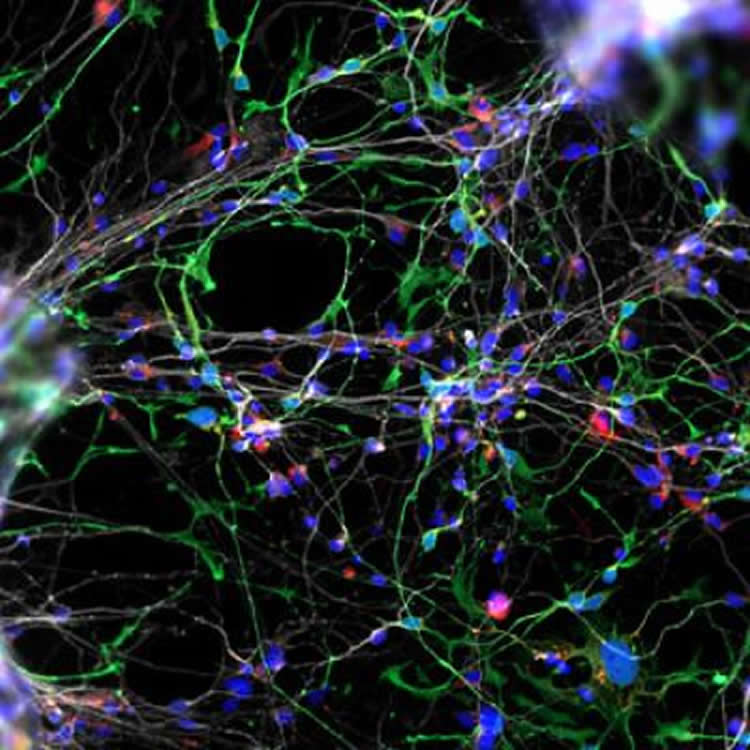
Cultured WS neurons have a distinct morphology. They are more arborized (treelike, with many dendritic branches) than neurons derived from typically developing individuals. “At the functional level, they make more synapses or connections to other neurons than what you would expect,” said Muotri. “That might underlie the WS super-social aspect and their gregarious human brain, giving insights into autism and other disorders that affect the social brain.”
The neuronal morphology was confirmed using a rare collection of WS postmortem brain tissue by Katerina Semendeferi, PhD, co-senior author and professor at UC San Diego Department of Anthropology. “One striking observation was that these cortical neurons in WS individuals are more complex than controls (typically developing children of same age). The morphological alterations that presumably appeared during WS gestation are kept postnatally.”
Muotri noted that the research represents one of the first efforts to use iPSCs and brain in-a-dish technology to generate novel insights about a disease process and not simply replicate data from other models.
But beyond that, he believes studying WS may help explain what makes humans social beings – a key development in the evolution of humanity. “It was our social power that made us a collaborative species,” said Muotri, “capable of dramatic transformation of our environment by creating poetry, music and technology.”
Co-authors include: Thanathom Chailangkarn, UC San Diego and National Center for Genetic Engineering and Biotechnology, Pathum Thani, Thailand; Cleber A. Trujillo, Beatriz C. Freitas, Timothy T. Brown, Branka Hrvoj-Mihic, Lisa Stefanacci, M. Collin Ard, Kari L. Hanson, Sarah Romero, and Anders M. Dale, UC San Diego; Roberto H. Herai, UC San Diego and Pontifica Universidade Catolica do Parana, Brazil; Diana X. Yu, Maria C. N. Marchetto, Cedric Bardy, Lauren McHenry, Anna Järvinen, Yvonne M. Searcy, Michelle DeWitt, Wenny Wong, Philip Lai, and Fred Gage, Salk Institute for Biological Studies; Bob Jacobs, Colorado College; Li Dai, and Julie R. Korenberg, University of Utah.
Funding: Funding support for this research came, in part, from the California Institute for Regenerative Medicine, the National Institutes of Health (P01 NICHD033113, NIH Director’s New Innovator Award Program 1-DP2-OD006495-01, R01MH09753, R01MH103134, U19MH107367), NARSAD, Engmann Foundation, JPB Foundation, Helmsley Foundation and Royal Thai Government.
Source: Scott LaFee – UCSD
Image Source: This Neurosciencenews image is credited to UC San Diego Health.
Original Research: Abstract for “A human neurodevelopmental model for Williams syndrome” by Thanathom Chailangkarn, Cleber A. Trujillo, Beatriz C. Freitas, Branka Hrvoj-Mihic, Roberto H. Herai, Diana X. Yu, Timothy T. Brown, Maria C. Marchetto, Cedric Bardy, Lauren McHenry, Lisa Stefanacci, Anna Järvinen, Yvonne M. Searcy, Michelle DeWitt, Wenny Wong, Philip Lai, M. Colin Ard, Kari L. Hanson, Sarah Romero, Bob Jacobs, Anders M. Dale, Li Dai, Julie R. Korenberg, Fred H. Gage, Ursula Bellugi, Eric Halgren, Katerina Semendeferi and Alysson R. Muotri in Nature. Published online August 10 2016 doi:10.1038/nature19067
[cbtabs][cbtab title=”MLA”]UCSD. “Neurodevelopmental Model of Williams Syndrome Offers Insight Into Human Social Brain.” NeuroscienceNews. NeuroscienceNews, 10 August 2016.
<https://neurosciencenews.com/social-brain-williams-syndrome-4823/>.[/cbtab][cbtab title=”APA”]UCSD. (2016, August 10). Neurodevelopmental Model of Williams Syndrome Offers Insight Into Human Social Brain. NeuroscienceNews. Retrieved August 10, 2016 from https://neurosciencenews.com/social-brain-williams-syndrome-4823/[/cbtab][cbtab title=”Chicago”]UCSD. “Neurodevelopmental Model of Williams Syndrome Offers Insight Into Human Social Brain.” https://neurosciencenews.com/social-brain-williams-syndrome-4823/ (accessed August 10, 2016).[/cbtab][/cbtabs]
Abstract
A human neurodevelopmental model for Williams syndrome
Williams syndrome is a genetic neurodevelopmental disorder characterized by an uncommon hypersociability and a mosaic of retained and compromised linguistic and cognitive abilities. Nearly all clinically diagnosed individuals with Williams syndrome lack precisely the same set of genes, with breakpoints in chromosome band 7q11.23. The contribution of specific genes to the neuroanatomical and functional alterations, leading to behavioural pathologies in humans, remains largely unexplored. Here we investigate neural progenitor cells and cortical neurons derived from Williams syndrome and typically developing induced pluripotent stem cells. Neural progenitor cells in Williams syndrome have an increased doubling time and apoptosis compared with typically developing neural progenitor cells. Using an individual with atypical Williams syndrome we narrowed this cellular phenotype to a single gene candidate, frizzled 9 (FZD9). At the neuronal stage, layer V/VI cortical neurons derived from Williams syndrome were characterized by longer total dendrites, increased numbers of spines and synapses, aberrant calcium oscillation and altered network connectivity. Morphometric alterations observed in neurons from Williams syndrome were validated after Golgi staining of post-mortem layer V/VI cortical neurons. This model of human induced pluripotent stem cells fills the current knowledge gap in the cellular biology of Williams syndrome and could lead to further insights into the molecular mechanism underlying the disorder and the human social brain.
“A human neurodevelopmental model for Williams syndrome” by Thanathom Chailangkarn, Cleber A. Trujillo, Beatriz C. Freitas, Branka Hrvoj-Mihic, Roberto H. Herai, Diana X. Yu, Timothy T. Brown, Maria C. Marchetto, Cedric Bardy, Lauren McHenry, Lisa Stefanacci, Anna Järvinen, Yvonne M. Searcy, Michelle DeWitt, Wenny Wong, Philip Lai, M. Colin Ard, Kari L. Hanson, Sarah Romero, Bob Jacobs, Anders M. Dale, Li Dai, Julie R. Korenberg, Fred H. Gage, Ursula Bellugi, Eric Halgren, Katerina Semendeferi and Alysson R. Muotri in Nature. Published online August 10 2016 doi:10.1038/nature19067




