

Mutations in the presenilin-1 gene are the most common cause of inherited, early-onset forms of Alzheimer’s disease. In a new study, published in Neuron, scientists replaced the normal mouse presenilin-1 gene with Alzheimer’s-causing forms of the human gene to discover how these genetic changes may lead to the disorder. Their surprising results may transform the way scientists design drugs that target these mutations to treat inherited or familial Alzheimer’s, a rare form of the disease that affects approximately 1 percent of people with the disorder. The study was partially funded by the National Institute of Neurological Disorders and Stroke (NINDS), part of the National Institutes of Health.
For decades, it has been unclear exactly how the presenilin mutations cause Alzheimer’s disease. Presenilin is a component of an important enzyme, gamma secretase, which cuts up amyloid precursor protein into two protein fragments, Abeta40 and Abeta42. Abeta42 is found in plaques, the abnormal accumulations of protein in the brain which are a hallmark of Alzheimer’s. Numerous studies suggested that presenilin-1 mutations increased activity of gamma-secretase. Investigators have developed drugs that block gamma-secretase, but they have so far failed in clinical trials to halt the disease
The study led by Raymond Kelleher, M.D., Ph.D. and Jie Shen, Ph.D., professors of neurology at Harvard Medical School, Boston, provides a plot twist in the association of presenilin-1 mutations and inherited Alzheimer’s disease. Using mice with altered forms of the presenilin gene, Drs. Kelleher and Shen discovered that the mutations may cause the disease by decreasing, rather than increasing, the activity of gamma-secretase.
One of the presenilin mutations also caused impairment of memory circuits in the mouse brain and age-dependent death of neurons.
“The findings by Drs. Shen and Kelleher are a significant departure from conventional thinking that should open up exciting and creative new possibilities at all levels of research, from basic molecular mechanisms all the way to clinical intervention,” said Roderick Corriveau, Ph.D., program director at NINDS.
“This is a very striking example where we have mutations that inactivate gamma-secretase function and yet they trigger an array of features that resemble Alzheimer’s disease, notably synaptic and cognitive deficits as well as neurodegeneration,” said Dr. Kelleher.
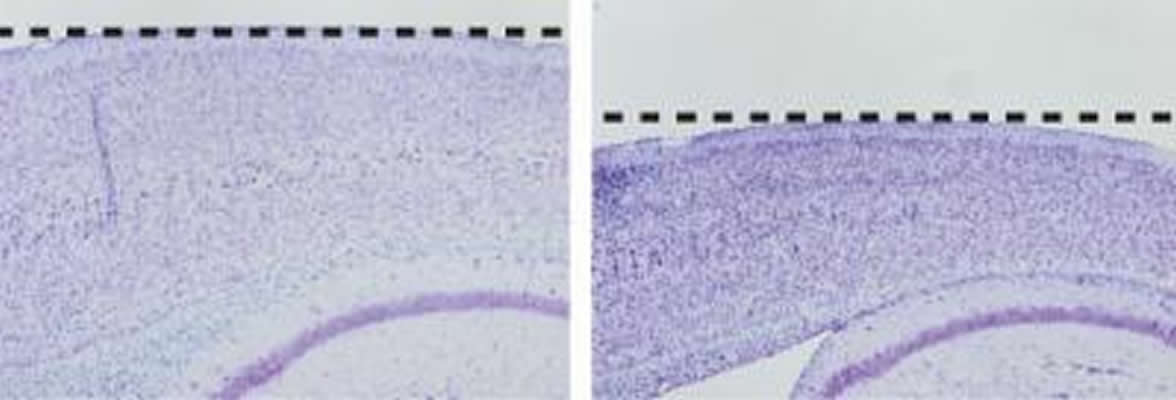
Although plaques are the main biological indicator of Alzheimer’s, neurodegenerative changes are also an important feature of the disease. These changes include loss of brain cells, accumulations of a protein called tau inside remaining neurons, cognitive deficits such as problems with memory, changes in the brain’s electrical activity and inflammation. Commonly used mouse models of the disease exhibit excessive plaque deposition, but do not show symptoms of neurodegeneration. According to Dr. Kelleher, this may be one reason that treatments developed in mice have not been successful in patients.
“This study is the first example of a mouse model in which a familial Alzheimer’s mutation is sufficient to cause neurodegeneration. The new model provides an opportunity that we hope will help with the development of therapies focusing on the devastating neurodegenerative changes that occur in the disease,” Dr. Kelleher said.
Dr. Shen’s previous work demonstrated that presenilins and gamma-secretase play an important role in learning and memory, communication between brain cells and neuronal survival, and cautioned against the use of gamma-secretase inhibitors for Alzheimer’s disease therapy. Later, a large phase III trial was stopped because treatment with a gamma-secretase inhibitor worsened the cognitive ability of patients.
Although the majority of cases are not inherited, familial Alzheimer’s disease is associated with early onset of the disorder, with symptoms often appearing before age 60. Drs. Shen and Kelleher hope that the mechanisms uncovered in this study may provide insight into the common forms of the disorder that affect more than five million people in the United States.
The results in this paper suggest a new approach for drug development. “We believe that restoring gamma-secretase would be a better, more effective therapeutic strategy for Alzheimer’s patients,” said Dr. Shen.
This work was supported by grants from the NINDS (NS041783, NS042818, NS075346), the Alzheimer’s Association, and the Pew Scholars Program in the Biomedical Sciences.
Contact: Press Office – NIH/NINDS
Source: NIH/NINDS press release
Image Source: The image is credited to Raymond Kelleher and Jie Shen, Harvard Medical School and is adapted from the NIH/NINDS press release
Original Research: Abstract for “Presenilin-1 Knockin Mice Reveal Loss-of-Function Mechanism for Familial Alzheimer’s Disease” by Dan Xia, Hirotaka Watanabe, Bei Wu, Sang Hun Lee, Yan Li, Evgeny Tsvetkov, Vadim Y. Bolshakov, Jie Shen, and Raymond J. Kelleher III in Neuron. Published online February 4 2015 doi:10.1016/j.neuron.2015.02.010




