Summary: Study identifies specific chemical features of tau that may cause it to accumulate in the brain and trigger Alzheimer’s disease.
Source: Tokyo Metropolitan University
Researchers from Tokyo Metropolitan University have discovered that a specific chemical feature of a key protein known as tau may cause it to accumulate in the brain and trigger illnesses like Alzheimer’s.
They found that disulfide bonds on certain amino acids act to stabilize tau and cause it to accumulate, an effect that got worse with increased oxidative stress. The identification of chemical targets triggering tau accumulation may lead to breakthrough treatments.
The tau protein is key to the healthy function of biological cells. It helps form and stabilize microtubules, the thin filaments that crisscross cell interiors to help keep them structurally rigid and provide ‘highways’ to shuttle molecules between organelles.
However, when they are not formed correctly, they can accumulate and form sticky clumps. In the brain, these aggregates block the firing of neurons and cause a wide range of neurodegenerative diseases known as tauopathies, one of which is Alzheimer’s disease. It is vastly important that scientists find the ‘switch’ that transforms tau from an indispensable part of cell function to a deadly pathology.
A team led by Associate Professor Kanae Ando of Tokyo Metropolitan University has been using model organisms like the Drosophila fruit fly to uncover how specific features of the tau protein cause it to stop working properly.
Flies can be genetically altered to express the same tau protein as in humans. By systematically modifying parts of the gene encoding for tau, they have been trying to pinpoint how certain features of mutant tau proteins affect their behavior.
In their most recent work, they found that alterations to amino acid residues in the protein known as cysteines in two different locations (C291 and C322) had a drastic effect on the amount and toxicity of tau. In a further breakthrough, the team pinned down the specific chemical feature responsible for making them toxic to normal cell function, that is, disulfide bonds formed by these cysteine groups.
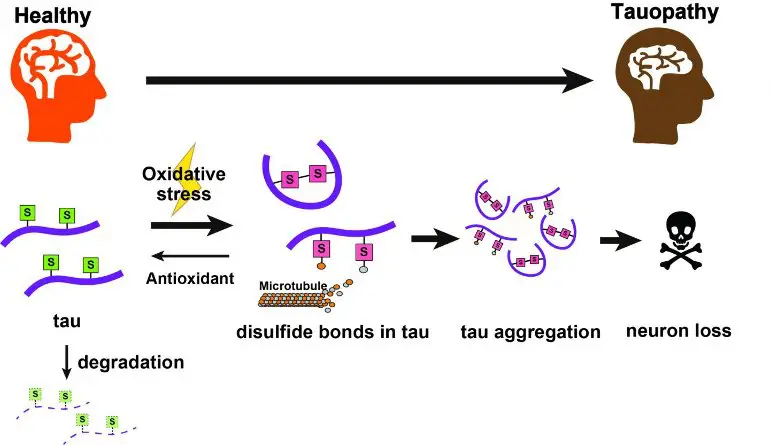
The toxic accumulation of tau got worse when cells were put in an environment with elevated levels of reactive oxygen species, as thiol groups on the cysteines were oxidized to form disulfide links. Biochemical environments with elevated oxidative stress are similar to those seen in patients with tauopathies. The co-expression of antioxidants to counter this effect helped natural processes clear away tau proteins, resulting in dramatically lower tau levels.
The team hope that knowledge of exactly which chemical groups are responsible for tau toxicity may lead to novel therapies which reduce or prevent tau accumulation, helping sufferers of tauopathies around the world.
Funding: This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) [JSPS KAKENHI Grant number 17H05703], a research award from the Hoan-sha Foundation, the Takeda Science Foundation, a research award from the Japan Foundation for Aging and Health, a Grant-in-Aid for Scientific Research on Challenging Research (Exploratory) [JSPS KAKENHI Grant number 309 19K21593], and a Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) [JSPS KAKENHI Grant number 26117004].
About this Alzheimer’s disease research news
Source: Tokyo Metropolitan University
Contact: Go Totsukawa – Tokyo Metropolitan University
Image: The image is credited to Tokyo Metropolitan University
Original Research: Closed access.
“Disulfide bond formation in microtubule-associated tau protein promotes tau accumulation and toxicity in vivo” by Kanae Ando et al. Human Molecular Genetics
Abstract
Disulfide bond formation in microtubule-associated tau protein promotes tau accumulation and toxicity in vivo
Accumulation of microtubule-associated tau protein is thought to cause neuron loss in a group of neurodegenerative diseases called tauopathies. In diseased brains, tau molecules adopt pathological structures that propagate into insoluble forms with disease-specific patterns. Several types of posttranslational modifications in tau are known to modulate its aggregation propensity in vitro, but their influence on tau accumulation and toxicity at the whole-organism level has not been fully elucidated.
Herein, we utilized a series of transgenic Drosophila models to compare systematically the toxicity induced by five tau constructs with mutations or deletions associated with aggregation, including substitutions at seven disease-associated phosphorylation sites (S7A and S7E), deletions of PHF6 and PHF6* sequences (ΔPHF6 and ΔPHF6*), and substitutions of cysteine residues in the microtubule binding repeats (C291/322A). We found that substitutions and deletions resulted in different patterns of neurodegeneration and accumulation, with C291/322A having a dramatic effect on both tau accumulation and neurodegeneration.
These cysteines formed disulfide bonds in mouse primary cultured neurons and in the fly retina, and stabilized tau proteins. Additionally, they contributed to tau accumulation under oxidative stress. We also found that each of these cysteine residues contributes to the microtubule polymerization rate and microtubule levels at equilibrium, but none of them affected tau binding to polymerized microtubules.
Since tau proteins expressed in the Drosophila retina are mostly present in the early stages of tau filaments self-assembly, our results suggest that disulfide bond formation by these cysteine residues could be attractive therapeutic targets.