

Energy’s Oak Ridge National Laboratory and the University of Tennessee have for the first time successfully characterized the earliest structural formation of the disease type of the protein that causes Huntington’s disease. The incurable, hereditary neurological disorder is always fatal and affects one in 10,000 Americans.
Huntington’s disease is caused by a renegade protein “huntingtin” that destroys neurons in areas of the brain concerned with the emotions, intellect and movement. All humans have the normal huntingtin protein, which is known to be essential to human life, although its true biological functions remain unclear.
Christopher Stanley, a Shull Fellow in the Neutron Scattering Science Division at ORNL, and Valerie Berthelier, a UT Graduate School of Medicine researcher who studies protein folding and misfolding in Huntington’s, have used a small-angle neutron scattering instrument, called Bio-SANS, at ORNL’s High Flux Isotope Reactor to explore the earliest aggregate species of the protein that are believed to be the most toxic.
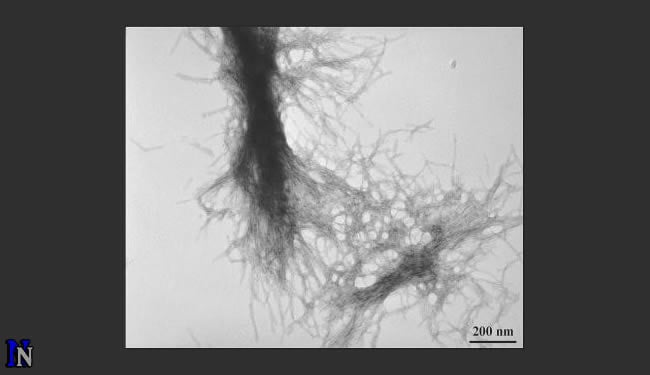
Stanley and Berthelier, in research published today in Biophysical Journal, were able to determine the size and mass of the mutant protein structures―from the earliest small, spherical precursor species composed of two (dimers) and three (trimers) peptides―along the aggregation pathway to the development of the resulting, later-stage fibrils. They were also able to see inside the later-stage fibrils and determine their internal structure, which provides additional insight into how the peptides aggregate.
“Bio-SANS is a great instrument for taking time-resolved snapshots. You can look at how this stuff changes as a function of time and be able to catch the structures at the earliest of times,” Stanley said. “When you study several of these types of systems with different glutamines or different conditions, you begin to learn more and more about the nature of these aggregates and how they begin forming.”
Normal huntingtin contains a region of 10 to 20 glutamine amino acids in succession. However, the DNA of Huntington’s disease patients encodes for 37 or more glutamines, causing instability in huntingtin fragments that contain this abnormally long glutamine repeat. Consequentially, the mutant protein fragment cannot be degraded normally and instead forms deposits of fibrils in neurons.
Those deposits, or clumps, were originally seen as the cause of the devastation that ensues in the brain. More recently researchers think the clumping may actually be a kind of biological housecleaning, an attempt by the brain cells to clean out these toxic proteins from places where they are destructive. Stanley and Berthelier set out to learn through neutron scattering what the toxic proteins were and when and where they occurred.
At the HFIR Bio-SANS instrument, the neutron beam comes through a series of mirrors that focus it on the sample. The neutrons interact with the sample, providing data on its atomic structure, and then the neutrons scatter, to be picked up by a detector. From the data the detector sends of the scattering pattern, researchers can deduce at a scale of less than billionths of a meter the size and shape of the diseased, aggregating protein, at each time-step along its growth pathway.
SANS was able to distinguish the small peptide aggregates in the sample solution from the rapidly forming and growing larger aggregates that are simultaneously present. In separate experiments, they were able to monitor the disappearance of the single peptides, as well as the formation of the mature fibrils.
Now that they know the structures, the hope is to develop drugs that can counteract the toxic properties in the early stages, or dissuade them from taking the path to toxicity. “The next step would be, let’s take drug molecules and see how they can interact and affect these structures,” Stanley said.
For now, the researchers believes Bio-SANS will be useful in the further study of Huntington’s disease aggregates and applicable for the study of other protein aggregation processes, such as those involved in Alzheimer’s and Parkinson’s diseases.
“That is the future hope. Right now, we feel like we are making a positive contribution towards that goal,” Stanley said.
Notes about this Huntington’s disease research article
The research was supported by the National Institutes of Health. HFIR and Bio-SANS are supported by the DOE Office of Science.
ORNL is managed by UT-Battelle for the Department of Energy’s Office of Science.
Author: Agatha Bardoel – Oak Ridge National Laboratory
Contact: Bill Cabage – DOE/Oak Ridge National Laboratory
Source: DOE/Oak Ridge National Laboratory press release
Image Source: Image adapted from ONL press release image





