Summary: A new collaborative study has helped researchers to uncover some of the earliest molecular events in ALS.
Source: Francis Crick Institute.
As the old adage goes, ‘two heads are better than one’. With the development of new technologies and increasingly specialist expertise, ground-breaking science needs to be a team effort.
But it isn’t always easy for researchers to work together. Finding the right people to collaborate with can be tricky, especially when some are understandably protective of their ideas.
Then there is the practical challenge of meeting up, exchanging ideas and carrying out the research, miles – and even time zones – apart.
A new study from Crick researchers shows that collaboration can be easy when you are part of a culture that supports it. We caught up with the scientists involved to find out how working together helped them uncover the earliest events in motor neuron disease.
The mystery
Three years ago, a group of clinical neurologists, molecular biologists and computer scientists from different London institutes decided to work together to solve the mystery of why motor neurons die in patients with amyotrophic lateral sclerosis (ALS), also known as motor neuron disease.
As a clinical neurologist, Rickie Patani sees first-hand the impact that ALS has on his patients.
“It’s a really devastating disease,” he says. “Patients progressively lose the ability to move, eat, speak and ultimately breathe.
“We set out to uncover the molecular events that lead to ALS, in the hope that one day we can develop new treatments for patients.”
The suspect
Previous studies had implicated deregulation of RNA – a molecule closely related to DNA that has a vital role in coding, decoding, regulating and expressing genes – in ALS. For instance, patients with a hereditary form of ALS often have genetic mutations that prevent their RNA from functioning properly.
But even with RNA expert Jernej Ule on board, comparing RNA sequencing in healthy and diseased motor neurons couldn’t provide the full picture.
Turning back the clock
Using cutting-edge stem cell technology, scientists in Rickie’s lab took skin cells from healthy volunteers and patients with ALS and turned them into stem cells capable of becoming many other cell types.
Then, using specific chemical signals, they ‘guided’ the stem cells into becoming motor neurons that they could study in the lab.
“By turning back the clock, we could watch what happened to the motor neurons over time to lead to the disease,” says Giulia Tyzack, a researcher in Rickie’s lab. “It was really amazing!”
Digging for treasure
Armed with a whole load of RNA sequencing data from healthy and diseased motor neurons at different stages of disease progression, Jernej and Rickie turned to Nick Luscombe and Raphaelle Luisier to drill down into the data and work out exactly what was going wrong. Nick and Raphaelle are bioinformaticians; highly skilled scientists who develop advanced computational techniques to study biological data.
“Initially, using conventional analysis, we didn’t detect any differences in RNA sequencing between healthy and diseased motor neurons,” says Raphaelle. “But we knew something must have been going wrong to make the ALS motor neurons die, so we wrote a new program to dig deeper into the genetic code – and when the results came back, we knew we were on to something.”
The analysis unearthed what was going wrong in ALS motor neurons. Parts of the RNA sequence that don’t code for proteins are usually cut out before the RNA is translated into protein, but in the ALS motor neurons this wasn’t happening as effectively. This guided the team to collectively discover that a protein called SFPQ, which normally resides inside the cell nucleus, was in fact leaving the nucleus in diseased motor neurons.
“It was like one big treasure hunt,” says Nick. “We had the map, and knew where we were looking, and with enough digging we found the gold!”
Cracking the case
The team had uncovered these molecular hallmarks inside human stem cell models of hereditary ALS. They next confirmed that animal models of hereditary ALS also shared the same features. But to see if the same events could explain non-hereditary forms of the disease, they looked at post-mortem spinal cord tissue from patients.
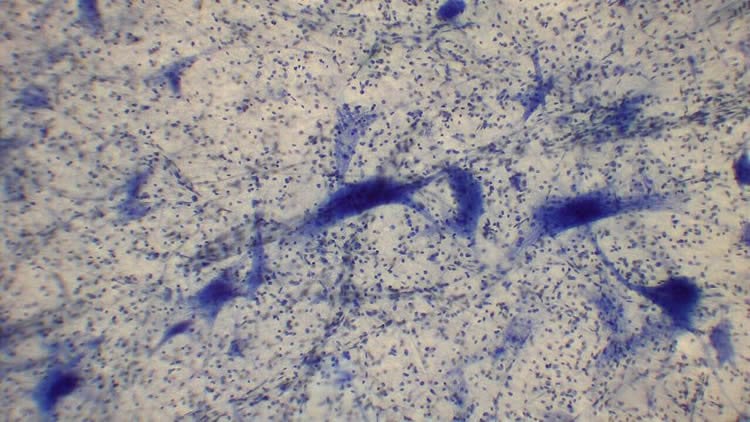
They found that the loss of SFPQ protein was consistent across the board, whether they looked at cells, mouse models or post-mortem tissue confirming that they had discovered an important molecular hallmark of ALS.
“Now that we know these key events are linked to motor neuron death in people with ALS, we can start to think about how we could develop new ways to detect and treat the disease,” says Rickie.
Under one roof
This project started before the Francis Crick Institute opened its doors to scientists in the summer of 2016.
For the first couple of years, the team were worked together across different sites, meeting up to share ideas when they could.
But since spring 2017, this scientific ‘dream team’ have all come together under one roof here at the Crick.
“It’s unbelievable how much of a difference it made all being two minutes’ walk from each other,” says Jernej. “The project was going well even when we were working in different institutes in London, but being able to chat to each other almost every day speeds things up dramatically. We finished the project within a year, while it might have taken two years or more if we weren’t all here at the Crick.”
By combining cellular models of motor neuron development, measurements of protein-RNA interactions and detailed statistical analysis, this diverse team of Crick scientists have shed light on potential causes of ALS, opening new opportunities to intervene and develop treatments.
This discovery goes to show that when it comes to science, two heads (or more) really are better than one.
Source: Greta Keenan – Francis Crick Institute
Publisher: Organized by NeuroscienceNews.com.
Image Source: NeuroscienceNews.com image is credited to 乌拉跨氪 licensed CC BY-SA 4.0.
Original Research: Open access research for “Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS” by Raphaelle Luisier, Giulia E. Tyzack, Claire E. Hall, Jamie S. Mitchell, Helen Devine, Doaa M. Taha, Bilal Malik, Ione Meyer, Linda Greensmith, Jia Newcombe, Jernej Ule, Nicholas M. Luscombe & Rickie Patani in Nature Communications. Published May 22 2018.
doi:10.1038/s41467-018-04373-8
[cbtabs][cbtab title=”MLA”]Francis Crick Institute “Researchers Shed New Light on Motor Neuron Death.” NeuroscienceNews. NeuroscienceNews, 25 May 2018.
<https://neurosciencenews.com/motor-neuron-death-9141/>.[/cbtab][cbtab title=”APA”]Francis Crick Institute (2018, May 25). Researchers Shed New Light on Motor Neuron Death. NeuroscienceNews. Retrieved May 25, 2018 from https://neurosciencenews.com/motor-neuron-death-9141/[/cbtab][cbtab title=”Chicago”]Francis Crick Institute “Researchers Shed New Light on Motor Neuron Death.” https://neurosciencenews.com/motor-neuron-death-9141/ (accessed May 25, 2018).[/cbtab][/cbtabs]
Abstract
Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS
Mutations causing amyotrophic lateral sclerosis (ALS) strongly implicate ubiquitously expressed regulators of RNA processing. To understand the molecular impact of ALS-causing mutations on neuronal development and disease, we analysed transcriptomes during in vitro differentiation of motor neurons (MNs) from human control and patient-specific VCP mutant induced-pluripotent stem cells (iPSCs). We identify increased intron retention (IR) as a dominant feature of the splicing programme during early neural differentiation. Importantly, IR occurs prematurely in VCP mutant cultures compared with control counterparts. These aberrant IR events are also seen in independent RNAseq data sets from SOD1- and FUS-mutant MNs. The most significant IR is seen in the SFPQ transcript. The SFPQ protein binds extensively to its retained intron, exhibits lower nuclear abundance in VCP mutant cultures and is lost from nuclei of MNs in mouse models and human sporadic ALS. Collectively, we demonstrate SFPQ IR and nuclear loss as molecular hallmarks of familial and sporadic ALS.