

Large, international NIH-supported study uses precision medicine to tackle neurological disorders.
Scientists searched the chromosomes of more than 4,000 Huntington’s disease patients and found that DNA repair genes may determine when the neurological symptoms begin. Partially funded by the National Institutes of Health, the results may provide a guide for discovering new treatments for Huntington’s disease and a roadmap for studying other neurological disorders.
“Our hope is to find ways that we can slow or delay the onset of Huntington’s devastating symptoms,” said James Gusella, Ph.D., director of the Center for Human Genetic Research at Massachusetts General Hospital (MGH), Boston, and the corresponding author of the study published online in Cell. “This could be possible because we now have a list of clinically proven genetic factors that influence the disease.”
Huntington’s disease is an inherited neurodegenerative disorder caused by mutations in a gene that encodes a protein called Huntingtin. Symptoms usually begin in midlife and include uncontrolled movements, emotional disturbances and, eventually, dementia. Although studies in humans and animals have discovered clues as to how the disorder works, there are no effective treatments.
For this study, the scientists tried a slightly different approach. They employed a novel application of Genome Wide Association Study analysis, a technique that scientists typically use to search for single letter changes to the DNA code on patients’ chromosomes, which may increase or decrease their chances of having the disease. Here, the scientists already knew the disease-causing gene and so they used the technique to search for other changes that determine when the disease starts. They found that at least three sites were associated with symptoms appearing earlier or later than expected, two on chromosome 15 and one on chromosome eight.
“This approach could have a significant impact on Huntington’s disease patients and researchers,” said Margaret Sutherland, Ph.D., program director, the National Institute of Neurological Disorders and Stroke, part of NIH. “It’s an example of how precision medicine may be applied to neurological disorders.”
The study was conducted by the Genetic Modifiers of Huntington’s Disease Consortium, an international team of scientists devoted to finding treatments for the disorder. Starting more than three decades ago they collected DNA samples and clinical information from patients, mainly in the United States, Canada and Europe.
“We thank the patients and their families for their commitment to research,” said Dr. Gusella. “It takes a dedicated global effort to meet the complex challenges presented by neurological disorders like Huntington’s disease.”
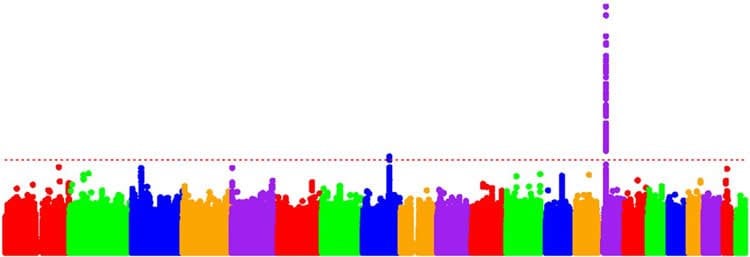
The scientists used the patients’ DNA and clinical information to study the age when movement problems began. Located on chromosome four, the Huntingtin gene is characterized by three letter repeats in the DNA code, called CAG-repeats. Disease-causing versions of the Huntingtin gene contain more than 35 CAG-repeats, which is higher than the six to 34 repeats found in normal versions. The greater number of CAGs a patient has, the greater the chances that symptoms will appear earlier in life. Focusing on patients who had 40 to 55 CAGs, the scientists found that some developed the disease earlier than expected while others developed it later.
The scientists then analyzed the patients’ chromosomes using gene chips that contain sites known for having single letter changes to the DNA code in normal human populations. After multiple rounds of searching they found that two sites on chromosome 15 were strongly associated with disease onset. One site was associated with hastening the disease by, on average, just over six years; another site was associated with slowing the disease by about a year and a half. They also found that a site on chromosome eight was associated with earlier disease onset by nearly one and a half years.
Although the scientists did not identify specific genes, they used several types of genome network analyses to show that disease onset may be controlled by genes that repair DNA, catalyze essential chemical reactions in cells, and assist with the division of mitochondria. In addition, they noted that a site within the code for MLH1, a DNA repair gene located on chromosome three, may be involved. Finally, the scientists showed that the sites on chromosomes 15 and eight may be associated with the beginning of psychiatric and cognitive problems caused by the disease.
“These results are an important step toward developing new treatments for Huntington’s disease. They help us understand how, through evolution, nature has learned to modify the disease-causing effects of Huntington’s disease mutations,” said Dr. Gusella. “As we study more patients we hope the information we obtain will soon reduce their suffering.”
Funding: This work was supported by grants from NIH (NS082079, NS091161, NS016367, HG002449, HG006074), the Medical Research Council (UK; G0801418, MR/L010305/1) and the CHDI Foundation.
Source: Christopher G. Thomas – NIH/NINDS
Image Credit: The image is credited to Gusella lab, MGH, Boston.
Original Research: Abstract for “Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease” by Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium in Cell. Published online June 18 2015 doi:10.1016/j.cell.2015.07.003
Abstract
Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease
Highlights
•GWA signals reveal loci that modify the age at onset of Huntington’s disease
•Effects at the chr15 locus hasten or delay onset by 6 or 1.4 years, respectively
•A single effect at the chr8 locus hastens onset by 1.6 years
•MLH1 association & pathway analysis implicate DNA handling in disease modification
Summary
As a Mendelian neurodegenerative disorder, the genetic risk of Huntington’s disease (HD) is conferred entirely by an HTT CAG repeat expansion whose length is the primary determinant of the rate of pathogenesis leading to disease onset. To investigate the pathogenic process that precedes disease, we used genome-wide association (GWA) analysis to identify loci harboring genetic variations that alter the age at neurological onset of HD. A chromosome 15 locus displays two independent effects that accelerate or delay onset by 6.1 years and 1.4 years, respectively, whereas a chromosome 8 locus hastens onset by 1.6 years. Association at MLH1 and pathway analysis of the full GWA results support a role for DNA handling and repair mechanisms in altering the course of HD. Our findings demonstrate that HD disease modification in humans occurs in nature and offer a genetic route to identifying in-human validated therapeutic targets in this and other Mendelian disorders.
“Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease” by Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium in Cell. Published online June 18 2015 doi:10.1016/j.cell.2015.07.003




