

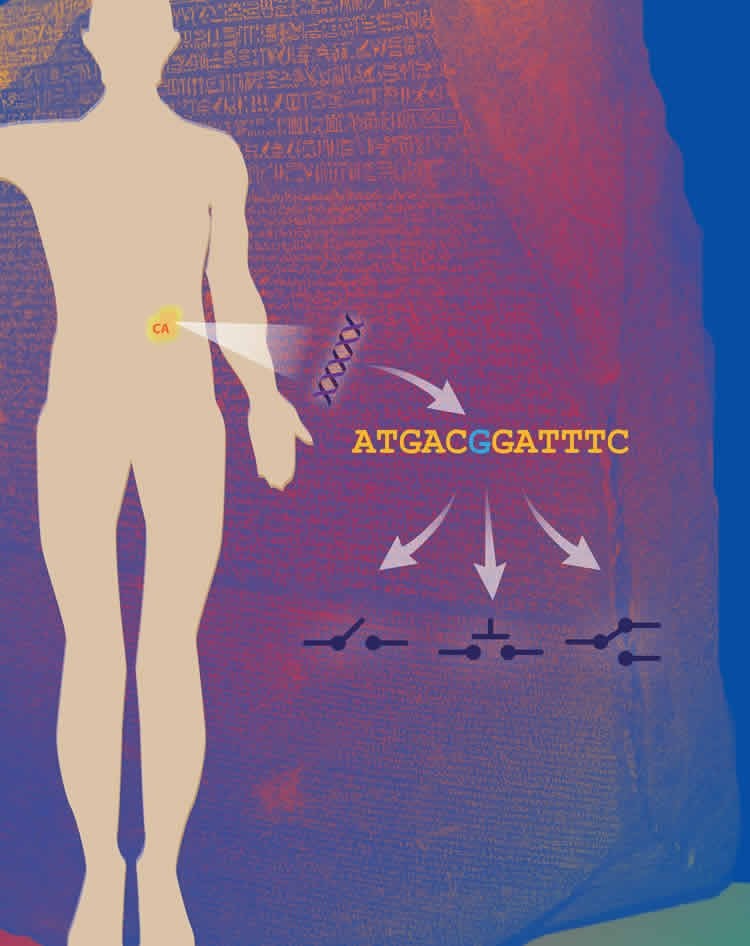
Scientists have discovered how genetic cancer mutations systematically attack the networks controlling human cells, knowledge critical for the future development of personalized precision cancer treatments.
Since the human genome was decoded more than a decade ago, cancer genomics studies have dominated life science worldwide and have been extremely successful at identifying mutations in individual patients and tumors. However, using this knowledge to develop improved cancer therapies has been severely hampered by the inability of researchers to link the mutations in genes to their corresponding proteins, the targets of most pharmaceutical drugs.
Translating DNA mutations
Researchers from the universities of Copenhagen, Yale, Zurich, Rome and Tottori have, in two landmark studies published in today’s CELL, unraveled how disease mutations target and damage the protein signaling networks within human cells on an unprecedented scale. The team has developed novel software that allows researchers to computationally translate the effects of cancer mutations on the function of proteins in individual patients.
Lead researcher on the projects, Prof Dr Rune Linding from the Biotech Research & Innovation Centre (BRIC) at the University of Copenhagen (UCPH), states: “The identification of distinct changes within our tissues that help predict and treat cancer is a major step forward and we are confident it can aid in the development of novel therapies and screening techniques.”
First author Dr Pau Creixell explains, “Given the tremendous amount and growth of genomic knowledge, a key challenge scientists face is how to interpret these data. This new software that can reveal how single DNA mutations can have dramatic molecular effects in cells by affecting critical enzymes called kinases.”
The studies, published back-to-back in CELL, demonstrate that kinases are not only simply switched ‘on’ or ‘off’ by cancer mutations but can also disturb other proteins and thereby drive normal cells to a more cancerous state.
Advancing personalized and tumor-specific medicine
Collaborator Dr Ben Turk, Associate Prof of Pharmacology at Yale University, adds that ”identifying mutations that effect the way that kinases regulate other proteins helps us to prioritize potential therapeutic targets, facilitating the advance of personalized medicine.”
It is becoming increasingly apparent that the genetic basis for each tumor is subtly different. This realization has led to healthcare centers spending millions of dollars sequencing individual patients and their tumors with the aim of utilizing this patient specific information to develop tailored, personalized therapies, with much greater efficacy. It is hoped that the novel tools described in these studies can provide much needed assistance to clinicians and researchers worldwide in interpreting this data.
Cancer biologist and co-author Dr Janine Erler states that “Studies like these are vital in enabling us to better understand the behavior of tumors in both individual and groups of patients, with these twin-papers we have seriously changed gears in the fight against cancer and other complex diseases.”
Funding: The work was supported by the European Research Council (ERC), the Lundbeck Foundation and Human Frontier Science Program.
Source: Anne Rahbek-Damm – BRIC
Image Source: The image is credited to Lori Waters, Waters Biomedical, 2015
Original Research: Full open access research for “Unmasking Determinants of Specificity in the Human Kinome” by Pau Creixell, Antonio Palmeri, Chad J. Miller, Hua Jane Lou, Cristina C. Santini, Morten Nielsen, Benjamin E. Turk, Rune Linding in Cell. Published online September 17 2015 doi:10.1016/j.cell.2015.08.057
Full open access research for “Kinome-wide Decoding of Network Attacking Mutations Rewiring ” by Pau Creixell, Erwin M. Schoof, Craig D. Simpson, James Longden, Chad J. Miller, Hua Jane Lou, Lara Perryman, Thomas R. Cox, Nevena Zivanovic, Antonio Palmeri, Agata Wesolowska-Andersen, Manuela Helmer-Citterich, Jesper Ferkinghoff-Borg, Hiroaki Itamochi, Bernd Bodenmiller, Janine T. Erler, Benjamin E. Turk, and Rune Linding in Cell. Published online September 17 2015 doi:10.1016/j.cell.2015.08.056
Abstract
Unmasking Determinants of Specificity in the Human Kinome
Highlights
•Residues driving specificity in the kinase and SH2 domains are globally identified
•Three new such residues, termed αC1, αC3, and APE-7, are experimentally validated
•Specificity and catalytic activity appear to be encoded in distinct sets of residues
•The global identification of determinants allows the modeling of rewiring mutations
Summary
Protein kinases control cellular responses to environmental cues by swift and accurate signal processing. Breakdowns in this high-fidelity capability are a driving force in cancer and other diseases. Thus, our limited understanding of which amino acids in the kinase domain encode substrate specificity, the so-called determinants of specificity (DoS), constitutes a major obstacle in cancer signaling. Here, we systematically discover several DoS and experimentally validate three of them, named the αC1, αC3, and APE-7 residues. We demonstrate that DoS form sparse networks of non-conserved residues spanning distant regions. Our results reveal a likely role for inter-residue allostery in specificity and an evolutionary decoupling of kinase activity and specificity, which appear loaded on independent groups of residues. Finally, we uncover similar properties driving SH2 domain specificity and demonstrate how the identification of DoS can be utilized to elucidate a greater understanding of the role of signaling networks in cancer
“Unmasking Determinants of Specificity in the Human Kinome” by Pau Creixell, Antonio Palmeri, Chad J. Miller, Hua Jane Lou, Cristina C. Santini, Morten Nielsen, Benjamin E. Turk, Rune Linding in Cell. Published online September 17 2015 doi:10.1016/j.cell.2015.08.057
Abstract
Kinome-wide Decoding of Network Attacking Mutations Rewiring
Highlights
•Mutations perturbing signaling networks are systematically classified and interpreted
•Several such functional mutations are identified in cancer and experimentally validated
•The results suggest that a single point mutant can have profound signaling effects
•Systematic interpretation of genomic data may assist future precision-medicine efforts
Summary
Cancer cells acquire pathological phenotypes through accumulation of mutations that perturb signaling networks. However, global analysis of these events is currently limited. Here, we identify six types of network-attacking mutations (NAMs), including changes in kinase and SH2 modulation, network rewiring, and the genesis and extinction of phosphorylation sites. We developed a computational platform (ReKINect) to identify NAMs and systematically interpreted the exomes and quantitative (phospho-)proteomes of five ovarian cancer cell lines and the global cancer genome repository. We identified and experimentally validated several NAMs, including PKCγ M501I and PKD1 D665N, which encode specificity switches analogous to the appearance of kinases de novo within the kinome. We discover mutant molecular logic gates, a drift toward phospho-threonine signaling, weakening of phosphorylation motifs, and kinase-inactivating hotspots in cancer. Our method pinpoints functional NAMs, scales with the complexity of cancer genomes and cell signaling, and may enhance our capability to therapeutically target tumor-specific networks.
“Kinome-wide Decoding of Network Attacking Mutations Rewiring ” by Pau Creixell, Erwin M. Schoof, Craig D. Simpson, James Longden, Chad J. Miller, Hua Jane Lou, Lara Perryman, Thomas R. Cox, Nevena Zivanovic, Antonio Palmeri, Agata Wesolowska-Andersen, Manuela Helmer-Citterich, Jesper Ferkinghoff-Borg, Hiroaki Itamochi, Bernd Bodenmiller, Janine T. Erler, Benjamin E. Turk, and Rune Linding in Cell. Published online September 17 2015 doi:10.1016/j.cell.2015.08.056




