A mechanism that alters the regulation of calcium in cells could be involved in neurodegenerative disease.
The calcium ion Ca2+ regulates an enormous number of cellular processes. Control of Ca2+ is therefore crucial and is achieved by precise regulation of proteins that allow the ion to move between different parts of the cell. Research led by Katsuhiko Mikoshiba and Kozo Hamada from the Laboratory for Developmental Neurobiology at the RIKEN Brain Science Institute has now revealed a previously unknown mechanism that regulates the release of Ca2+ from stores inside cells and which could be involved in several brain disorders.
The research team investigated the role of an enzyme called transglutaminase type 2 (TG2) in the regulation of the inositol 1,4,5-trisphosphate receptor (IP3R) protein, which forms a channel in the membranes of cellular compartments. The IP3R channel opens and closes in response to regulatory signals to control the release of Ca2+ from cellular stores.
“IP3R is required for vital processes that involve calcium signaling, but excessive IP3R function promotes cell death processes, so regulation is important,” says Mikoshiba. “TG2 levels are higher than normal in various neurodegenerative diseases, including Alzheimer’s, Huntington’s and Parkinson’s diseases, so we were interested in the effect of TG2 on IP3R.”
The researchers measured the amount of Ca2+ that passed through IP3Rs in a simplified system of cellular compartments that were isolated from mouse cells and loaded with calcium. Adding TG2 to this system reduced the amount of Ca2+ that could pass through IP3Rs. In living cells, blocking the function of TG2 had the reverse effect: it increased Ca2+ release from the cellular stores. Both findings show that TG2 blocks the function of IP3Rs.
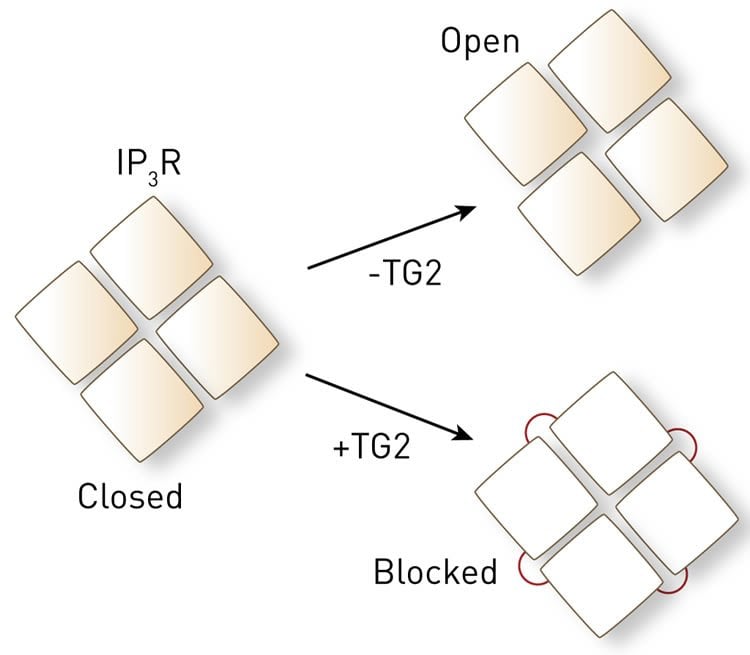
The team then went further and looked at exactly how TG2 affects IP3Rs on a molecular level. “Four IP3R proteins assemble to form a calcium channel, which normally opens by changing the spatial relationship between the four proteins,” explains Mikoshiba. “TG2 links the four proteins together and chronically locks the spatial relationship between IP3R proteins to block their function.”
Because TG2 levels are abnormal in many neurodegenerative disorders, the researchers also investigated how TG2 affects IP3Rs in a mouse model of Huntington’s disease. They found that reduced IP3R function in mice with Huntington’s disease occurred only in association with elevated TG2, suggesting that increased TG2 levels contribute to the disease.
“This mechanism of TG2 might serve as a general principle for many other diseases of the brain and other tissues in which TG2 is upregulated by disease-inducing agents,” says Mikoshiba.
Contact: Katsuhiko Mikoshiba – RIKEN
Source: RIKEN press release
Image Source: The image is credited to Katsuhiko Mikoshiba and is adapted from the RIKEN press release
Original Research: Abstract for “Aberrant calcium signaling by transglutaminase-mediated posttranslational modification of inositol 1,4,5-trisphosphate receptors” by Kozo Hamada, Akiko Terauchi, Kyoko Nakamura, Takayasu Higo, Nobuyuki Nukina, Nagisa Matsumoto, Chihiro Hisatsune, Takeshi Nakamura, and Katsuhiko Mikoshiba in PNAS. Published online September 27 2014 doi:10.1073/pnas.1409730111