Summary: Study reports protein aggregates within cells are cleared out by the endocytosis process. The findings could help with the development of new treatments for ALS.
Source: University of Arizona
A therapeutic intervention for amyotrophic lateral sclerosis, better known as ALS or Lou Gehrig’s disease, could be on the horizon thanks to unexpected findings by University of Arizona researchers.
ALS is the progressive degeneration of motor neurons that causes people to lose the ability to move and eventually speak, eat and breathe.
Within the neuronal cells of patients with ALS and other neurodegenerative diseases, two proteins – TDP-43 and FUS – are often found in bundles of molecular junk called aggregates, which can accumulate to deadly levels.
“It’s not clear yet if TDP-43 aggregates themselves are truly toxic or a sign that things have gotten really bad in a cell, and this is its last Hail Mary trying to keep things in order,” said Ross Buchan, assistant professor of molecular and cellular biology and a member of the BIO5 Institute. “These aggregates could possibly be toxic because they are trapping other useful molecules and not letting them do their job.”
Buchan and his team set out to investigate how healthy cells clear harmful aggregates from the cell.
What they found was that the aggregates were being removed via endocytosis, which was surprising for two reasons. First, the textbook definition of endocytosis is a process in which proteins, nutrients and chemical signals from outside the cell are brought inside to be degraded and recycled by the lysosome. But in this case, endocytosis was working on aggregates that were already inside the cell. And second, there’s already a mechanism, called autophagy, in place for recycling junk that originated from within a cell, yet endocytosis was doing what autophagy should have been doing instead.
“Autophagy – and also likely, although it’s still uncertain, endocytosis – often slows as we age, and there are genes that are mutated in that pathway that are associated with some neurodegenerative diseases. So people thought the reason aggregates form when we get old, or when you have these diseases, is because that pathway isn’t working very well,” Buchan said.
Additionally, the accumulation of aggregates slows the endocytosis pathway further, creating a negative feedback loop within the cell.
“If we genetically or chemically impede the pathway, then the TDP-43 protein accumulates and becomes super toxic. The cool thing, as far as a therapy for ALS is concerned, is that we can also do the reverse,” Buchan said. “We can make the endocytosis pathway work better by over-expressing parts of it, like putting the gas pedal down so it goes really fast. When we do that, then the TDP-43 aggregates are cleared really efficiently and it’s no longer toxic.”
Many of the paper’s experiments were performed in yeast cells, but the general findings are likely translatable to human cells based on initial findings. Buchan called yeast “a powerful genetic tool,” for understanding cellular processes, including those in human disease.
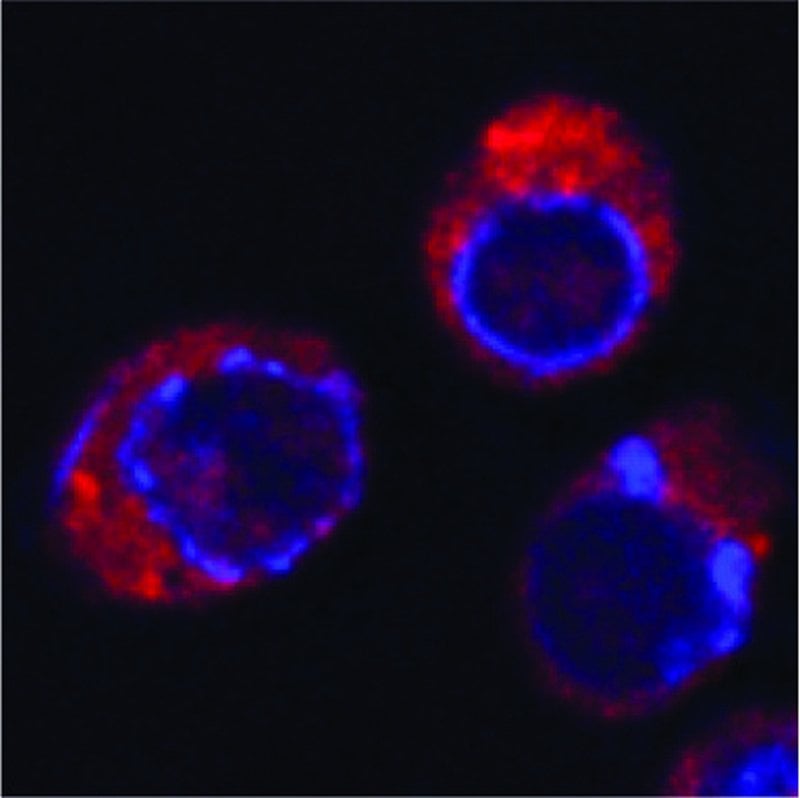
While the results from Buchan’s lab are unexpected – “If I were to pull a textbook off the self, it would say endocytosis is for things that are outside the cell, not inside, so it’s still pretty heretical,” he said – there are other labs with data suggesting endocytosis can also clear already internalized proteins.
The next step is to determine how TPD-43 and FUS enter the endocytic pathway, and then to develop ways to make endocytosis work better in these cells.
“There are genetic ways to do that, but not chemically at the moment,” Buchan said. “We think if we have a drug that inhibits the negative regulators of endocytosis, the pathway will go faster as a result. We have a couple ideas of where to start next.”
The findings were published in the journal Molecular and Cellular Biology. Buchan’s co-authors include undergraduate Amanda Warner, former post-doctoral fellow Guangbo Liu, graduate student Aaron Byrd, former graduate student Fen Pei, assistant research scientist Eman Basha and former lab technician Allison Buchanan.
Source:
University of Arizona
Media Contacts:
Mikayla Mace – University of Arizona
Image Source:
The image is credited to Ross Buchan.
Original Research: Closed access
“Cdc48/VCP and Endocytosis Regulate TDP-43 and FUS Toxicity and Turnover”. Guangbo Liu, Aaron Byrd, Amanda N. Warner, Fen Pei, Eman Basha, Allison Buchanan, J. Ross Buchan.
Molecular and Cellular Biology doi:10.1128/MCB.00256-19.
Abstract
Cdc48/VCP and Endocytosis Regulate TDP-43 and FUS Toxicity and Turnover
Amyotrophic lateral sclerosis (ALS) is a fatal motor neuron degenerative disease. TDP-43 (TAR DNA-binding protein 43) and FUS (fused in sarcoma) are aggregation-prone RNA-binding proteins that in ALS can mislocalize to the cytoplasm of affected motor neuron cells, often forming cytoplasmic aggregates in the process. Such mislocalization and aggregation are implicated in ALS pathology, though the mechanism(s) of TDP-43 and FUS cytoplasmic toxicity remains unclear. Recently, we determined that the endocytic function aids the turnover (i.e., protein degradation) of TDP-43 and reduces TDP-43 toxicity. Here, we identified that Cdc48 and Ubx3, a Cdc48 cofactor implicated in endocytic function, regulates the turnover and toxicity of TDP-43 and FUS expressed in Saccharomyces cerevisiae. Cdc48 physically interacts and colocalizes with TDP-43, as does VCP, in ALS patient tissue. In yeast, FUS toxicity also depends strongly on endocytic function but not on autophagy under normal conditions. FUS expression also impairs endocytic function, as previously observed with TDP-43. Taken together, our data identify a role for Cdc48/VCP and endocytic function in regulating TDP-43 and FUS toxicity and turnover. Furthermore, endocytic dysfunction may be a common defect affecting the cytoplasmic clearance of ALS aggregation-prone proteins and may represent a novel therapeutic target of promise.