Study led by National University of Singapore scientists reveals new insights on how neurons tag and transport key metabolic enzymes; Findings may enhance understanding of rare motor disorder.
An international team of researchers, led by scientists at the National University of Singapore (NUS), has identified a protein that regulates the growth of neurons by transporting key metabolic enzymes to the tips of neural cells. Their findings, published on 14 September 2015, in Developmental Cell, a leading journal in the field of developmental biology, open up new avenues for design of drugs for ataxia, a motor coordination disorder.
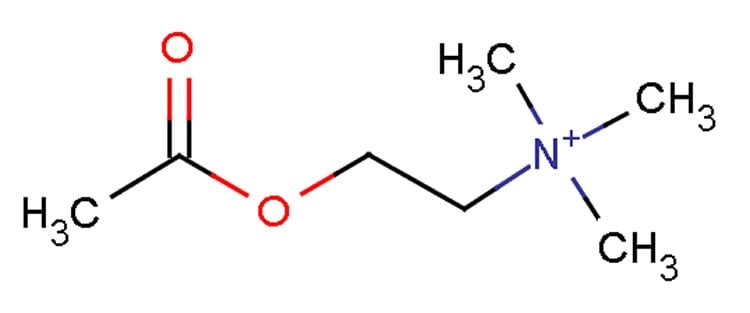
Neurotransmitters — chemicals used by brain cells to communicate — are essential for brain function. In particular, acetylcholine, which was the first neurotransmitter to be discovered, is involved in cognition and motor functions. Although much is known about the synthesis and secretion of this critical neurotransmitter, the spatial and temporal regulation of acetylcholine synthesis remains unclear. Specifically, how key metabolic enzymes such as ATP citrate lyase (ACL) and choline acetyltransferase (ChAT) find their way to the right region of the neuron is largely unknown.
To unravel this puzzle, the NUS team, led by Associate Professor Boon Chuan Low and his postdoctoral fellow Dr Jichao Sun, from the Department of Biological Sciences and Mechanobiology Institute at NUS, collaborated with researchers from the Yong Loo Lin School of Medicine at NUS and the University of Michigan (U-M). They identified and characterised a protein that transports the enzyme ACL to the tips of neurons, where it subsequently recruits another enzyme ChAT for acetylcholine synthesis. This ACL-transporting protein, called BNIP-H, was first linked to Cayman ataxia, a rare genetic disorder affecting a region of the brain involved in motor control and which leads to difficulty in coordinating complex movements, by Professor Margit Burmeister of U-M.
The research team looked at the biological roles of BNIP-H in cell lines, primary neuron cultures and zebrafish using molecular genetics, protein biochemistry and high speed imaging. They found that BNIP-H acts as a tag, marking ACL for transport by the enzyme kinesin-1 to the neuron terminals. Once there, BNIP-H and ACL synergistically recruit ChAT, triggering the targeted release of acetylcholine. Using mass spectrometry, the researchers showed that expressing more BNIP-H in cultured cells could increase acetylcholine secretion while knockdown of BNIP-H reduced acetylcholine secretion. The BNIP-H-induced increase of acetylcholine in turn launches a positive feedback loop involving the MAPK/ERK signalling pathway that ultimately promotes growth of neurites, which are projections from neurons.
“BNIP-H defines the precise localisation, duration and strength of acetylcholine signalling that determines the growth of neurons and the coordination of body movements,” explained Assoc Prof Low, the corresponding author of the paper.
The study also provides the first experimental data solidifying the link between dysfunctional cholinergic (acetylcholine) secretion and Cayman ataxia. The researchers showed that a BNIP-H mutant associated with Cayman ataxia caused defects in the transport of the ACL enzyme. Furthermore, they could also reproduce motor dysfunctions of Cayman ataxia in zebrafish by knocking down BNIP-H, ACL or ChAT enzymes. Interestingly, the lack of BNIP-H could be ‘rescued’ by the addition of a cholinergic agonist, suggesting that the loss of acetylcholine secretion resulting from BNIP-H mutation could explain some of the symptoms of Cayman ataxia.
Said Assoc Prof Low, “We established the first ACL-based ataxia model in the zebrafish that recapitulates the ataxic phenotype seen in human patients. Our findings provide the first detailed understanding at the molecular, cellular and organism levels on how defects in ACL trafficking impairs cholinergic signalling that leads to the development of ataxia.”
Moving forward, the authors hope to further characterise the role of BNIP-H in cholinergic neurotransmission. Their work also serves as a foundation for further studies into acetylcholine-related diseases, and may lead to new treatments that involve BNIP-H.
“Our findings could provide new direction to better understand causes of cholinergic-related diseases, such as Alzheimer’s disease, Down’s syndrome, ataxia and schizophrenia. Changing the activity of BNIP-H or/and its downstream effectors might be used to treat those diseases caused by dysregulation of cholinergic neurotransmission,” said Assoc Prof Low.
Source: Carolyn Fong – National University of Singapore
Image Source: The image is in the public domain
Original Research: Abstract for “BNIP-H Recruits the Cholinergic Machinery to Neurite Terminals to Promote Acetylcholine Signaling and Neuritogenesis” by Jichao Sun, Catherine Qiurong Pan, Ti Weng Chew, Fengyi Liang, Margit Burmeister, and Boon Chuan Low in Developmental Cell. Published online September 3 2015 doi:10.1016/j.devcel.2015.08.006
Abstract
BNIP-H Recruits the Cholinergic Machinery to Neurite Terminals to Promote Acetylcholine Signaling and Neuritogenesis
Highlights
•BNIP-H acts as a scaffold to link and transport ACL on kinesin-1 toward neurite ends
•BNIP-H and ACL recruit ChAT at neurite ends to increase ACh level for neuritogenesis
•Human Cayman ataxia splice mutant impairs trafficking of ACL and neurite outgrowth
•Bnip-h depletion disrupts zebrafish ACh signal, motor neuron development, and mobility
Summary
Synthesis and release of neurotransmitters such as acetylcholine (ACh) are key to synaptic function. However, little is known about the spatial regulation of their synthesizing machinery. Here, we demonstrate that ataxia-related protein BNIP-H/Caytaxin links kinesin-1 (KLC1) to ATP citrate lyase (ACL), a key enzyme for ACh synthesis, and transports it toward neurite terminals. There, BNIP-H/ACL complex synergistically recruits another enzyme choline acetyltransferase (ChAT), leading to enhanced secretion of ACh. ACh then activates MAPK/ERK via muscarinic receptors to promote neurite outgrowth. In mice deficient in BNIP-H, ACL fails to interact with KLC1, and formation of the ACL/ChAT complex is prevented, whereas the disease-associated BNIP-H mutation fails to target ACL for neurite outgrowth. Significantly, Bnip-h knockdown in zebrafish causes developmental defect in motor neurons through impaired cholinergic pathway, leading to motor disorder. Therefore, precise targeting of the cholinergic machinery through BNIP-H is essential for the local production of ACh for morphogenesis and neurotransmission.
“BNIP-H Recruits the Cholinergic Machinery to Neurite Terminals to Promote Acetylcholine Signaling and Neuritogenesis” by Jichao Sun, Catherine Qiurong Pan, Ti Weng Chew, Fengyi Liang, Margit Burmeister, and Boon Chuan Low in Developmental Cell. Published online September 3 2015 doi:10.1016/j.devcel.2015.08.006