Summary: A new study reports that abnormalities in the SOD1 protein are a common factor in all types of motor neuron diseases.
Source: University of Sydney
Researchers have found an abnormal protein usually linked to a rare inherited form of motor neuron disease is present in all types of motor neuron disease, suggesting a common link between the different forms of the disease.
The study, published in the neuroscience journal Brain, is the first to confirm toxic changes to the protein in individuals with genetic or non-genetic forms of motor neuron disease.
Amyotrophic lateral sclerosis (ALS) is the most common form of motor neuron disease. Ten percent of ALS cases are hereditary, with remaining cases lacking an apparent genetic cause.
“The results suggest this abnormal protein contributes to cell death in many forms of motor neuron disease, not just rare genetic cases of motor neuron disease,” says senior author Professor Kay Double from the Brain and Mind Center, Faculty of Medicine and Health.
“It is a big step in advancing our understanding of motor neuron disease. Our findings will direct further research and could ultimately lead to more effective treatments.”
Normally, the protein superoxide dismutase 1 (SOD1) protects cells, but a mutation in its gene is thought to make the protein “toxic”; this toxic protein form is associated with hereditary forms of ALS. Abnormal mutant SOD1 is only found in regions of the spinal cord where nerve cells die, implicating this abnormal protein in cell death.
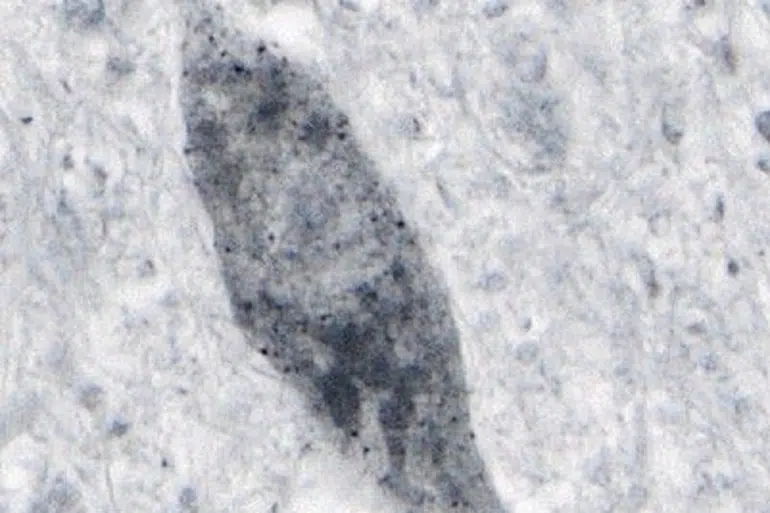
Previous investigations into the role of toxic forms of SOD1 protein largely focused on mutant forms of the protein and were primarily conducted using animal and cellular models of ALS.
The study, led by a team from the University of Sydney’s Brain and Mind Center, advances our understanding of the causes of motor neuron disease by studying this abnormal protein in post-mortem tissues from patients with ALS.
“We have shown for the first time that mechanisms of disease long hypothesized to occur in animal and cellular models are present in patients with motor neuron disease,” says lead author Dr. Benjamin Trist from the Brain and Mind Center, Faculty of Medicine and Health.
“This is a significant milestone in our understanding of ALS and motor neuron disease more broadly.”
In related experiments, Professor Double and her team are also currently studying how abnormal SOD1 interacts with other disease-linked proteins in motor neuron disease. This work is in press and will be published in Acta Neuropathologica Communications.
About this motor neuron disease research news
Author: Press Office
Source: University of Sydney
Contact: Press Office – University of Sydney
Image: The image is credited to Trist et al.
Original Research: Open access.
“Altered SOD1 maturation and post-translational modification in amyotrophic lateral sclerosis spinal cord” by Benjamin G Trist et al. Brain
Abstract
Altered SOD1 maturation and post-translational modification in amyotrophic lateral sclerosis spinal cord
Aberrant self-assembly and toxicity of wild-type and mutant superoxide dismutase 1 (SOD1) has been widely examined in silico, in vitro, and in transgenic animal models of amyotrophic lateral sclerosis (ALS). Detailed examination of the protein in disease-affected tissues from ALS patients, however, remains scarce.
We employed histological, biochemical and analytical techniques to profile alterations to SOD1 protein deposition, subcellular localization, maturation and post-translational modification in post-mortem spinal cord tissues from ALS cases and controls. Tissues were dissected into ventral and dorsal spinal cord grey matter to assess the specificity of alterations within regions of motor neuron degeneration.
We provide evidence of the mislocalization and accumulation of structurally-disordered, immature SOD1 protein conformers in spinal cord motor neurons of SOD1-linked and non-SOD1-linked familial ALS cases, and sporadic ALS cases, compared with control motor neurons. These changes were collectively associated with instability and mismetallation of enzymatically-active SOD1 dimers, as well as alterations to SOD1 post-translational modifications and molecular chaperones governing SOD1 maturation.
Atypical changes to SOD1 protein were largely restricted to regions of neurodegeneration in ALS cases, and clearly differentiated all forms of ALS from controls. Substantial heterogeneity in the presence of these changes was also observed between ALS cases.
Our data demonstrates that varying forms of SOD1 proteinopathy are a common feature of all forms of ALS, and support the presence of one or more convergent biochemical pathways leading to SOD1 proteinopathy in ALS. The majority of these alterations are specific to regions of neurodegeneration, and may therefore constitute valid targets for therapeutic development.