Summary: Diet-induced changes to the reward system and innate differences may predispose mice to over-eating.
Source: SfN
A combination of innate differences and diet-induced changes to the reward system may predispose some mice to overeat, according to research recently published in Journal of Neuroscience.
Food is fuel, but the rising levels of sugar and fat in modern diets make the brain treat it as a reward. One brain region called the ventral pallidum (VP) serves as a hub between reward areas and the hypothalamus, a region involved in feeding behavior. Intertwining food and reward can lead to overeating and may be a contributing factor to diet-induced obesity.
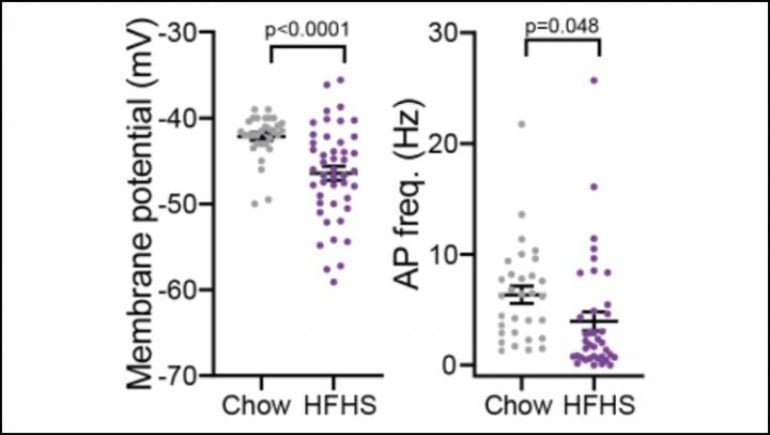
Gendelis, Inbar, et al. measured electrical activity in the VP of mice with unlimited access to high fat, high sugar food for several months. Eating the unhealthy diet changed the electrical properties of VP neurons: the membrane voltage and firing rate decreased, making it harder for neurons to send messages to each other. The change was more pronounced in the mice that gained the most weight.
The same set of electrical bursts strengthened synapses in weight gainers but weakened them in mice that gained the least weight.
These signaling differences may be innate and not caused by the unhealthy diet itself: the same plasticity differences appear between mice with natural high and low food-seeking behavior even without exposure to an unhealthy diet.
About this neuroscience research news
Source: SfN
Contact: Calli McMurray – SfN
Image: The image is credited to Gendelis, Inbar et al., JNeurosci 2020.
Original Research: Closed access.
“Metaplasticity in the Ventral Pallidum as a Potential Marker for the Propensity To Gain Weight in Chronic High-Calorie Diet” by Shani Gendelis, Dorrit Inbar, Kineret Inbar, Shanee Mesner and Yonatan M. Kupchik. Journal of Neuroscience
Abstract
Metaplasticity in the Ventral Pallidum as a Potential Marker for the Propensity To Gain Weight in Chronic High-Calorie Diet
A major driver of obesity is the increasing palatability of processed foods. Although reward circuits promote the consumption of palatable food their involvement in obesity remains unclear. The ventral pallidum (VP) is a key hub in the reward system that encodes the hedonic aspects of palatable food consumption and participates in various proposed feeding circuits. However, there is still no evidence for its involvement in developing diet-induced obesity. Here we examine, using male C57bl6/J mice and patch-clamp electrophysiology, how chronic high-fat-high-sugar (HFHS) diet changes the physiology of the VP and whether mice that gain the most weight differ in their VP physiology from others. We found that 10-12 weeks of HFHS diet hyperpolarized and decreased the firing rate of VP neurons without a major change in synaptic inhibitory input. Within the HFHS group, the top 33% weight gainers (WG) had a more hyperpolarized VP with longer latency to fire action potentials upon depolarization compared to bottom 33% weight gainers (NWG). Weight gainers also showed synaptic potentiation of inhibitory inputs both at the millisecond and minute ranges. Moreover, we found that the tendency to potentiate the inhibitory inputs to the VP might exist in overeating mice even before exposure to HFHS, thus making it a potential property of being an overeater. These data point to the VP as a critical player in obesity and suggest that hyperpolarized membrane potential of, and potentiated inhibitory inputs to, VP neurons may play a significant role in promoting overeating of palatable food.
SIGNIFICANCE STATEMENT:
In modern world, where highly-palatable food is readily available, overeating is often driven by motivational, rather than metabolic, needs. It is thus conceivable that reward circuits differ between obese and normal-weight individuals. But is such difference, if it exists, innate or develops with overeating? Here we reveal synaptic properties in the ventral pallidum, a central hub of reward circuits, that differ between mice that gain the most and the least weight when given unlimited access to highly-palatable food. We show that these synaptic differences exist also without exposure to palatable food, potentially making them innate properties that render some more susceptible than others to overeat. Thus, the propensity to overeat may have a strong innate component embedded in reward circuits.