

Discovery will aid development of treatments for this debilitating disease.
Scientists at the Montreal Neurological Institute and Hospital (MNI) have identified novel gene mutations that cause hereditary spastic paraplegia (HSP), a step forward in efforts to treat this debilitating disease.
It is estimated that between two and 10 people per 100,000 in the general population have HSP, a disease characterized by weakness or spasticity in the lower limbs. HSP is caused by mutations inherited from one or both parents.
While mutations in more than 70 genes are known or suspected to be responsible for HSP at the present time, many families remain undiagnosed, suggesting the causes of HSP are not entirely known.
MNI researchers are part of a Canadian consortium of scientists (CanHSP) that analyze the DNA of patients with HSP. Two years ago MNI researchers identified one family with autosomal recessive HSP that appeared to be caused by mutations in the gene CAPN1, previously unknown to be a factor in the disease. The scientists needed more examples of CAPN1 causing HSP symptoms to prove a causal relationship between the two.
A year ago, 20 families suffering from HSP were investigated, in collaboration with scientists from Morocco. The MNI researchers, led by Ziv Gan-Or, a postdoctoral fellow, found two families among them whose HSP originated from CAPN1 mutations.
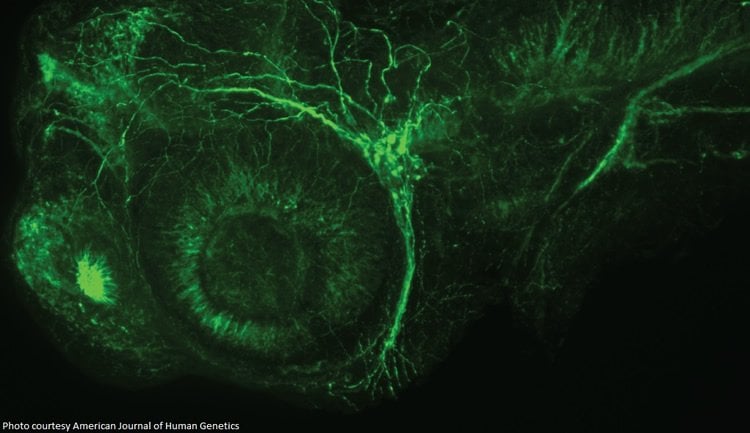
To test the link between the CAPN1 mutations and HSP, researchers created animal models using worms, flies, and zebrafish embryos, where the gene was “knocked down”; basically simulating the effect of the mutations. As a result of the knockdown, animal models showed characteristics similar to those experienced by HSP sufferers, proving a link between the mutations and HSP.
With this new information, researchers around the world can test people with HSP for the mutations, possibly resulting in diagnosis. While only one-to-two per cent of HSP may be caused by CAPN1 mutations, the discovery of their role in HSP improves neuroscience’s overall understanding of the disease and aids the search for drugs that can treat or cure HSP.
“The immediate impact will be to enable diagnosis of patients,” says Dr. Guy Rouleau, director of the MNI and one of the paper’s authors. “Treatment will follow, but is a more long-term project that will require some time.”
“Our finding will help physicians and genetic counselors as it will now be possible to add CAPN1 to the panel of genes examined when meeting with HSP patients,” says Bouchra Ouled Amar Bencheikh, a postdoctoral fellow with McGill University’s Faculty of Medicine, and one of the paper’s authors. “At the pathophysiological level, using three animal models with CAPN1 gene deficiency, we observed locomotor defects or neuronal disorganization depending on the model. Interestingly, similar observations were reported for the gene spastin, the most common genetic cause of HSP. The discovery of such a mechanism will help the development of new therapeutics for these disorders.”
Funding: This research was supported with funds from The Canadian Institutes for Health Research (CIHR).
Source: Shawn Hayward – McGill University
Image Source: The image is credited to American Journal of Human Genetics.
Original Research: Abstract for “Mutations in CAPN1 Cause Autosomal-Recessive Hereditary Spastic Paraplegia” by Ziv Gan-Or, Naima Bouslam, Nazha Birouk, Alexandra Lissouba, Daniel B. Chambers, Julie Vérièpe, Alaura Androschuck, Sandra B. Laurent, Daniel Rochefort, Dan Spiegelman, Alexandre Dionne-Laporte, Anna Szuto, Meijiang Liao, Denise A. Figlewicz, Ahmed Bouhouche, Ali Benomar, Mohamed Yahyaoui, Reda Ouazzani, Grace Yoon, Nicolas Dupré, Oksana Suchowersky, Francois V. Bolduc, J. Alex Parker, Patrick A. Dion, Pierre Drapeau, Guy A. Rouleau, and Bouchra Ouled Amar Bencheikh in American Journal of Human Genetics. Published online May 5 2016 doi:10.1016/j.ajhg.2016.04.002
Abstract
Mutations in CAPN1 Cause Autosomal-Recessive Hereditary Spastic Paraplegia
Hereditary spastic paraplegia (HSP) is a genetically and clinically heterogeneous disease characterized by spasticity and weakness of the lower limbs with or without additional neurological symptoms. Although more than 70 genes and genetic loci have been implicated in HSP, many families remain genetically undiagnosed, suggesting that other genetic causes of HSP are still to be identified. HSP can be inherited in an autosomal-dominant, autosomal-recessive, or X-linked manner. In the current study, we performed whole-exome sequencing to analyze a total of nine affected individuals in three families with autosomal-recessive HSP. Rare homozygous and compound-heterozygous nonsense, missense, frameshift, and splice-site mutations in CAPN1 were identified in all affected individuals, and sequencing in additional family members confirmed the segregation of these mutations with the disease (spastic paraplegia 76 [SPG76]). CAPN1 encodes calpain 1, a protease that is widely present in the CNS. Calpain 1 is involved in synaptic plasticity, synaptic restructuring, and axon maturation and maintenance. Three models of calpain 1 deficiency were further studied. In Caenorhabditis elegans, loss of calpain 1 function resulted in neuronal and axonal dysfunction and degeneration. Similarly, loss-of-function of the Drosophila melanogaster ortholog calpain B caused locomotor defects and axonal anomalies. Knockdown of calpain 1a, a CAPN1 ortholog in Danio rerio, resulted in abnormal branchiomotor neuron migration and disorganized acetylated-tubulin axonal networks in the brain. The identification of mutations in CAPN1 in HSP expands our understanding of the disease causes and potential mechanisms.
“Mutations in CAPN1 Cause Autosomal-Recessive Hereditary Spastic Paraplegia” by Ziv Gan-Or, Naima Bouslam, Nazha Birouk, Alexandra Lissouba, Daniel B. Chambers, Julie Vérièpe, Alaura Androschuck, Sandra B. Laurent, Daniel Rochefort, Dan Spiegelman, Alexandre Dionne-Laporte, Anna Szuto, Meijiang Liao, Denise A. Figlewicz, Ahmed Bouhouche, Ali Benomar, Mohamed Yahyaoui, Reda Ouazzani, Grace Yoon, Nicolas Dupré, Oksana Suchowersky, Francois V. Bolduc, J. Alex Parker, Patrick A. Dion, Pierre Drapeau, Guy A. Rouleau, and Bouchra Ouled Amar Bencheikh in American Journal of Human Genetics. Published online May 5 2016 doi:10.1016/j.ajhg.2016.04.002




