

Scientists at The Scripps Research Institute (TSRI) have discovered how one gene is essential to hearing, uncovering a cause of deafness and suggesting new avenues for therapies.
The new study, published November 20 in the journal Neuron, shows how mutations in a gene called Tmie can cause deafness from birth. Underlining the critical nature of their findings, researchers were able to reintroduce the gene in mice and restore the process underpinning hearing.
“This raises hopes that we could, in principle, use gene-therapy approaches to restore function in hair cells and thus develop new treatment options for hearing loss,” said Professor Ulrich Müller, senior author of the new study, chair of the Department of Molecular and Cellular Neuroscience and director of the Dorris Neuroscience Center at TSRI.
The Gene Responsible
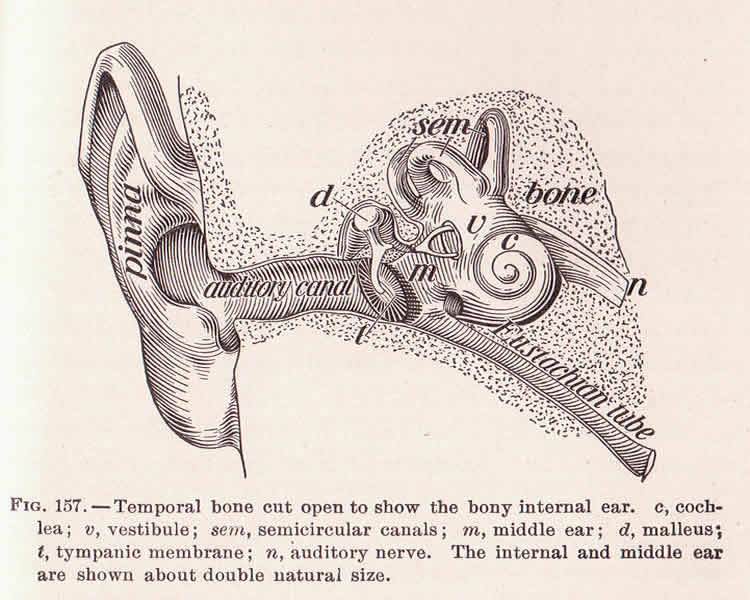
The ear is a complex machine that converts mechanical sound waves into electric signals for the brain to process. When a sound wave enters the ear, the uneven ends (stereocilia) of the inner ear’s hair cells are pushed back, like blades of grass bent by a heavy wind. The movement causes tension in the strings of proteins (tip links) connecting the stereocilia, which sends a signal to the brain through ion channels that run through the tips of the hair cell bundles.
This process of converting mechanical force into electrical activity, called mechanotransduction, still poses many mysteries. In this case, researchers were in the dark about how signals were passed along the tip links to the ion channels, which shape electrical signals.
To track down this unknown component, researchers in the new study built a library of thousands of genes with the potential to affect mechanotransduction.
The team spent six months screening the genes to see if the proteins the genes produced interacted with tip link proteins. Eventually, the team found a gene, Tmie, whose protein, TMIE, interacts with tip link proteins and connects the tip links to a piece of machinery near the ion channel.
A Path to New Treatments
This discovery answers a long-standing question in neuroscience. Scientists have long known that mutations in the Tmie gene could cause deafness—but they weren’t sure how.
Once they found that TMIE plays a role bridging the tip links and ion channels, the researchers bred a population of “knock-out mice” that lacked the gene. They examined the hair cells of the mice with electrophysiological techniques and found that without TMIE, no electrical signal could be evoked in hair cells after stimulation.
“The mechanotransduction current is gone; the mouse is totally deaf,” explained Bo Zhao, a research associate in the Müller lab and first author of the new paper.
In a second experiment, the researchers reintroduced Tmie to mice that had been deaf since birth and found the electrical signals were restored.
Müller said the next challenge is to find out how the individual components of the complex mechanotransduction system form in hair cells. “We would also like to understand the biophysical principles by which these proteins convert mechanical signals into electrical signals,” he said.
In addition to Müller and Zhao, the authors of the paper “TMIE is an Essential Component of the Mechanotransduction Machinery of Cochlear Hair Cells” were Zizhen Wu, Linxuan Yan, Wei Xiong and Sarah Harkins-Perry of TSRI; and Nicolas Grillet of Stanford University.
Funding for the research was provided by the National Institutes of Health (UM DC005965 and DC007704), the Dorris Neuroscience Center, the Skaggs Institute for Chemical Biology and the Bundy Foundation.
Contact: Office of Communications – Scripps Research Institute
Source: Scripps Research Institute press release
Image Source: The image is credited to Alvin Davison and is in the public domain
Original Research: Abstract for “TMIE Is an Essential Component of the Mechanotransduction Machinery of Cochlear Hair Cells” by Bo Zhao, Zizhen Wu, Nicolas Grillet, Linxuan Yan, Wei Xiong, Sarah Harkins-Perry, and Ulrich Müller in Neuron. Published online November 20 2014 doi:10.1016/j.neuron.2014.10.041




