

Summary: Mutations to the MARK4 gene causes the properties of tau to change, making them more likely to aggregate and become more insoluble.
Source: Tokyo Metropolitan University
Researchers from Tokyo Metropolitan University have discovered a new mechanism by which clumps of tau protein are created in the brain, killing brain cells and causing Alzheimer’s disease. A specific mutation to an enzyme called MARK4 changed the properties of tau, usually an important part of the skeletal structure of cells, making it more likely to aggregate, and more insoluble. Getting to grips with mechanisms like this may lead to breakthrough treatments.
Alzheimer’s disease is a life-changing, debilitating condition, affecting tens of millions of people worldwide. According to the World Health Organization, it is the most common cause of senile dementia, with numbers worldwide expected to double every 20 years if left unchecked.
Alzheimer’s is said to be caused by the build-up of tangled clumps of a protein called “tau” in brain cells. These sticky aggregates cause neurons to die, leading to impairment in memory and motor functions. It is not yet clear how and why tau builds up in the brain cells of Alzheimer’s patients. Understanding the cause and mechanism behind this unwanted clumping would open up the way to new treatments and ways to prevent the disease.
A team led by Associate Professor Kanae Ando of Tokyo Metropolitan University has been exploring the role played by the MARK4 (Microtubule Affinity Regulating Kinase 4) enzyme in Alzheimer’s disease. When everything is working normally, the tau protein is an important part of the structure of cells, or the cytoskeleton. To keep the arms of the cytoskeleton or microtubules constantly building and disassembling, MARK4 actually helps tau detach from the arms of this structure.
Problems start when a mutation occurs in the gene that provides the blueprint for making MARK4. Previous work had already associated this with an increased risk of Alzheimer’s, but it was not known why this was the case. The team artificially introduced mutations into transgenic drosophila fruit flies that also produce human tau, and studied how the proteins changed in vivo.
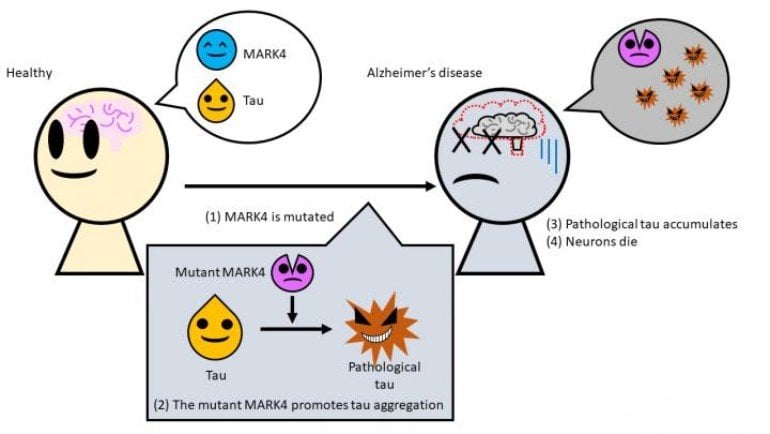
They discovered that this mutant form of MARK4 makes changes to the tau protein, creating a pathological form of tau. Not only did this “bad” tau have an excess of certain chemical groups that caused it to misfold, they found that it aggregated much more easily and were no longer soluble in detergents. This made it easier for tau to form the tangled clumps that causes neurons to degenerate.
MARK4 has also been found to cause a wide range of other diseases which involve the aggregation and buildup of other proteins. That’s why the team’s insights into tau protein buildup may lead to new treatments and preventative measures for an even wider variety of neurodegenerative conditions.
Funding: This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) [JSPS KAKENHI Grant number 17H05703], a research award from the Hoan-sha Foundation, the Takeda Science Foundation, a research award from the Japan Foundation for Aging and Health, a Grant-in-Aid for Scientific Research on Challenging Research (Exploratory) [JSPS KAKENHI Grant number 19K21593], and Research Funding for Longevity Science 19-7 from the National Center for Geriatrics and Gerontology, Japan.
About this genetics and Alzheimer’s disease research news
Source: Tokyo Metropolitan University
Contact: Go Totsukawa – Tokyo Metropolitan University
Image: The image is credited to Tokyo Metropolitan University
Original Research: Closed access.
“Microtubule Affinity Regulating Kinase 4 with an Alzheimer’s disease-related mutation promotes tau accumulation and exacerbates neurodegeneration” by Kanae Ando et al. Journal of Biological Chemistry
Abstract
Microtubule Affinity Regulating Kinase 4 with an Alzheimer’s disease-related mutation promotes tau accumulation and exacerbates neurodegeneration
Accumulation of the microtubule-associated protein tau is associated with Alzheimer’s disease (AD). In AD brain, tau is abnormally phosphorylated at many sites, and phosphorylation at Ser262 and Ser356 play critical roles in tau accumulation and toxicity. Microtubule-affinity regulating kinase 4 (MARK4) phosphorylates tau at those sites, and a double de novo mutation in the linker region of MARK4, ΔG316E317D, is associated with an elevated risk of AD. However, it remains unclear how this mutation affects phosphorylation, aggregation, and accumulation of tau and tau-induced neurodegeneration. Here, we report that MARK4ΔG316E317D increases the abundance of highly phosphorylated, insoluble tau species, and exacerbates neurodegeneration via Ser262/356-dependent and -independent mechanisms. Using transgenic Drosophila expressing human MARK4 (MARK4wt) or a mutant version of MARK4 (MARK4ΔG316E317D), we found that co-expression of MARK4wt and MARK4ΔG316E317Dincreased total tau levels and enhanced tau-induced neurodegeneration, and that MARK4ΔG316E317D had more potent effects than MARK4wt. Interestingly, the in vitrokinase activities of MARK4wt and MARK4ΔG316E317D were similar. When tau phosphorylation at Ser262 and Ser356 was blocked by alanine substitutions, MARK4wtdid not promote tau accumulation or exacerbate neurodegeneration, while co-expression of MARK4ΔG316E317D did. Both MARK4wt and MARK4ΔG316E317D increased the levels of oligomeric forms of tau; however, only MARK4ΔG316E317D further increased the detergent insolubility of tau in vivo. Together, these findings suggest that MARK4ΔG316E317D increases tau levels and exacerbates tau toxicity via a novel gain-of-function mechanism, and that modification in this region of MARK4 may impact disease pathogenesis.




