Summary: A new study shows the effectiveness of ketamine in treating depression in a mouse model of the disease and also helps bring together two hypotheses for the cause of depression.
Source: Penn State University.
New insight from depressed mice helps researchers unite two hypotheses.
New research demonstrates the effectiveness of ketamine to treat depression in a mouse model of the disease and brings together two hypotheses for the cause of depression. The research, led by Bernhard Lüscher, professor of biology and of biochemistry and molecular biology at Penn State University, is in press and will be published in the September 15, 2016, print edition of the journal Biological Psychiatry.
“Depression is the second most expensive health problem that we face worldwide, but this fact is not very well known because there is a stigma attached to depression and people don’t like to talk about it,” said Lüscher. “About 17 percent of Americans will be treated for depression at some point in their lives, but there are limited treatment options and about one-third of patients do not respond to these treatments.”
Lüscher and his colleagues generated a mouse model for depression by introducing a mutation into a gene that codes for one of the subunits of a receptor for GABA — the second most abundant chemical used by nerve cells in the brain to communicate. GABA functions mainly to reduce the activity of nerve cells. The receptor mutation results in a reduction in GABA signaling of about 15 to 20 percent and mimics reductions in GABA signaling seen in patients with depression. The mice that have the mutation exhibit traits associated with depression, such as reduced pleasure seeking, and they become normal again following treatment with antidepressant medications.
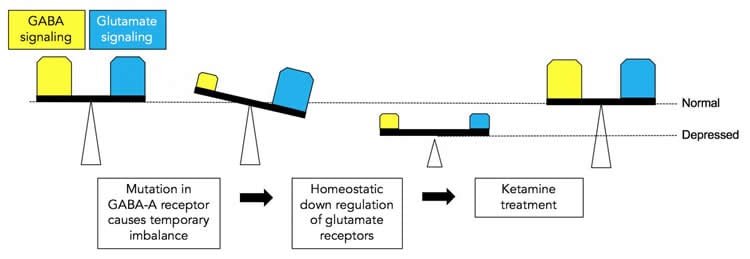
“You can think of GABA as acting like the brakes of a car — its function is to slow activity in nerve cells,” said Lüscher. “Its counterpart is glutamate, another signaling chemical in the brain that acts as the accelerator of nerve-cell activity. When we reduced the function of GABA in our mice, we were surprised to see that the level of glutamate was also reduced. This result suggests that the brain has mechanisms that maintain a balance between the brakes and the accelerator to prevent brain activity from going out of control, a state we refer to as homeostasis.”
The researchers treated the mice with low doses of ketamine, an experimental antidepressant drug known to act by transiently blocking a major class of glutamate receptors in nerve cells. “Treatment with ketamine not only normalized the behavior and brought glutamate receptor levels back up to normal in our mice, but GABA function also was restored,” said Lüscher. Importantly, the effects of ketamine were only observed in mice with the receptor mutation where nerve signaling was defective, and not in normal mice. “Our results bring together the hypothesis that depression results from deficits in GABA signaling and the hypothesis that depression results from deficits in glutamate signaling. We showed that the depression-like behavior in our mice results from the reduction of both GABA and glutamate, and importantly, that both can be restored with a single dose of ketamine.”
The researchers plan to use their mouse model to better understand how ketamine functions to develop safer alternatives. “Ketamine has many advantages over currently used antidepressant medications,” said Lüscher. “It acts quickly and has long lasting effects, but it is addictive and can induce psychosis, so we hope to use our model to better understand how ketamine works biochemically. We can then begin to develop ketamine-like drugs without the unwanted side effects.”
In addition to Luscher, the research team includes Zhen Ren, Sarah J. Jefferson, Matthew Shorey, and Thomas Fuchs at Penn State and Horia Pribiag and David Stellwagen at McGill University in Montreal, Canada.
Funding: The research was supported by the National Institutes of Mental Health (grant numbers MH089111 and MH099851), the Canadian Institutes of Heath Research, and the Natural Sciences and Engineering Research Council of Canada.
Source: Barbara K. Kennedy – Penn State University
Image Source: This NeuroscienceNews.com image is credited to Penn State University.
Video Source: The video is credited to PSU Science Media Relations and Public Information.
Original Research: Full open access research for “Bidirectional Homeostatic Regulation of a Depression-Related Brain State by Gamma-Aminobutyric Acidergic Deficits and Ketamine Treatment” by Zhen Ren, Horia Pribiag, Sarah J. Jefferson, Matthew Shorey, Thomas Fuchs, David Stellwagen, and Bernhard Luscher in Biological Psychiatry. Published online February 12 2016 doi:10.1016/j.biopsych.2016.02.009
[cbtabs][cbtab title=”MLA”]Penn State University. “How Depression and Antidepressant Drugs Work.” NeuroscienceNews. NeuroscienceNews, 18 May 2016.
<https://neurosciencenews.com/ketamine-depression-mouse-model-4252/>.[/cbtab][cbtab title=”APA”]Penn State University. (2016, May 18). How Depression and Antidepressant Drugs Work. NeuroscienceNews. Retrieved May 18, 2016 from https://neurosciencenews.com/ketamine-depression-mouse-model-4252/[/cbtab][cbtab title=”Chicago”]Penn State University. “How Depression and Antidepressant Drugs Work.” NeuroscienceNews.
https://neurosciencenews.com/ketamine-depression-mouse-model-4252/ (accessed May 18, 2016).[/cbtab][/cbtabs]
Abstract
Bidirectional Homeostatic Regulation of a Depression-Related Brain State by Gamma-Aminobutyric Acidergic Deficits and Ketamine Treatment
Background
Major depressive disorder is increasingly recognized to involve functional deficits in both gamma-aminobutyric acid (GABA)ergic and glutamatergic synaptic transmission. To elucidate the relationship between these phenotypes, we used GABAA receptor γ2 subunit heterozygous (γ2+/−) mice, which we previously characterized as a model animal with construct, face, and predictive validity for major depressive disorder.
Methods
To assess possible consequences of GABAergic deficits on glutamatergic transmission, we quantitated the cell surface expression of N-methyl-D-aspartate (NMDA)-type and alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)-type glutamate receptors and the function of synapses in the hippocampus and medial prefrontal cortex of γ2+/− mice. We also analyzed the effects of an acute dose of the experimental antidepressant ketamine on all these parameters in γ2+/− versus wild-type mice.
Results
Modest defects in GABAergic synaptic transmission of γ2+/− mice resulted in a strikingly prominent homeostatic-like reduction in the cell surface expression of NMDA-type and AMPA-type glutamate receptors, along with prominent functional impairment of glutamatergic synapses in the hippocampus and medial prefrontal cortex. A single subanesthetic dose of ketamine normalized glutamate receptor expression and synaptic function of γ2+/− mice to wild-type levels for a prolonged period, along with antidepressant-like behavioral consequences selectively in γ2+/− mice. The GABAergic synapses of γ2+/− mice were potentiated by ketamine in parallel but only in the medial prefrontal cortex.
Conclusions
Depressive-like brain states that are caused by GABAergic deficits involve a homeostatic-like reduction of glutamatergic transmission that is reversible by an acute, subanesthetic dose of ketamine, along with regionally selective potentiation of GABAergic synapses. The data merge the GABAergic and glutamatergic deficit hypotheses of major depressive disorder.
“Bidirectional Homeostatic Regulation of a Depression-Related Brain State by Gamma-Aminobutyric Acidergic Deficits and Ketamine Treatment” by Zhen Ren, Horia Pribiag, Sarah J. Jefferson, Matthew Shorey, Thomas Fuchs, David Stellwagen, and Bernhard Luscher in Biological Psychiatry. Published online February 12 2016 doi:10.1016/j.biopsych.2016.02.009